Combination immune checkpoint blockade (ICB) therapies have been successful in the clinic, but the exact mechanisms of their synergy are not completely understood. For example, co-inhibition of PD-1 and TIGIT can be superior to either alone, although the distinct contributions of each to antitumor immunity remain unclear. Recently reported in Immunity, Banta and Xu et al. provided a mechanistic view into the observed synergy between these pathways.
Previously, it was found that antibody blockade of CD226, a costimulatory T cell marker regulated by TIGIT, reduced the efficacy of dual anti-PD-L1 and anti-TIGIT treatment, indicating a relationship between PD-1 and CD226. Confirming this interplay, the authors found that early after implantation, CT26 colon carcinoma-bearing Cd226-/- mice, but not wild-type mice, were incapable of responding to either anti-PD-1 or anti-TIGIT monotherapy. In addition, CD8+ T cells from Cd226-/- mice showed inferior killing of antigen-expressing target cells ex vivo. The loss of CD226 ligands PVR/PVRL2 from target cells also reduced killing by wild-type CD8+ T cells, emphasizing a role of CD226 costimulation in CD8+ T cell functionality.
With the importance of CD226 in ICB response established in a mouse model, the researchers questioned whether CD226 may also be important in patient ICB efficacy. Analyzing bulk RNAseq data from three clinical trials of atezolizumab (anti-PD-L1) in non-small cell lung carcinoma (NSCLC), they found that CD226 expression correlated positively with overall and progression-free survival. The expression of other costimulatory genes, particularly CD28, PDCD1, and TIGIT, among others, did not display this correlation, suggesting a unique role of CD226 in the patient immunotherapy response.
Next, the team investigated the phenotypes of CD226- and CD28-expressing CD8+ T cells within NSCLC tumors using CyTOF. As expected, CD226 and CD28 were primarily expressed on T cells. Dividing these cells into four populations based on their CD226 and CD28 co-expression, differences in phenotypic marker expression were observed. Although the double-positive CD28+CD226+ population was the rarest, it expressed high CD27, associated with antigen-specific expansion and memory, and low TIM-3 and CD103, compared to the CD28- populations. The two CD28+ populations displayed a similar profile.
To analyze T cell phenotypes more rigorously, the authors used scRNAseq, investigating CD8+ T cells from NSCLC tumors and adjacent healthy tissue. Eight distinct CD8+ T cell clusters were uncovered, matching previously characterized phenotypes (e.g., effector, resident memory). In both tumors and healthy tissue, CD226 and CD28 were variably expressed among these distinct clusters. Interestingly, CD226 and CD28 were most strongly co-expressed in proliferative (MKI67+) and cytotoxic memory (KLRB1+) CD8+ T cell states, likely to be involved in immunotherapy response.
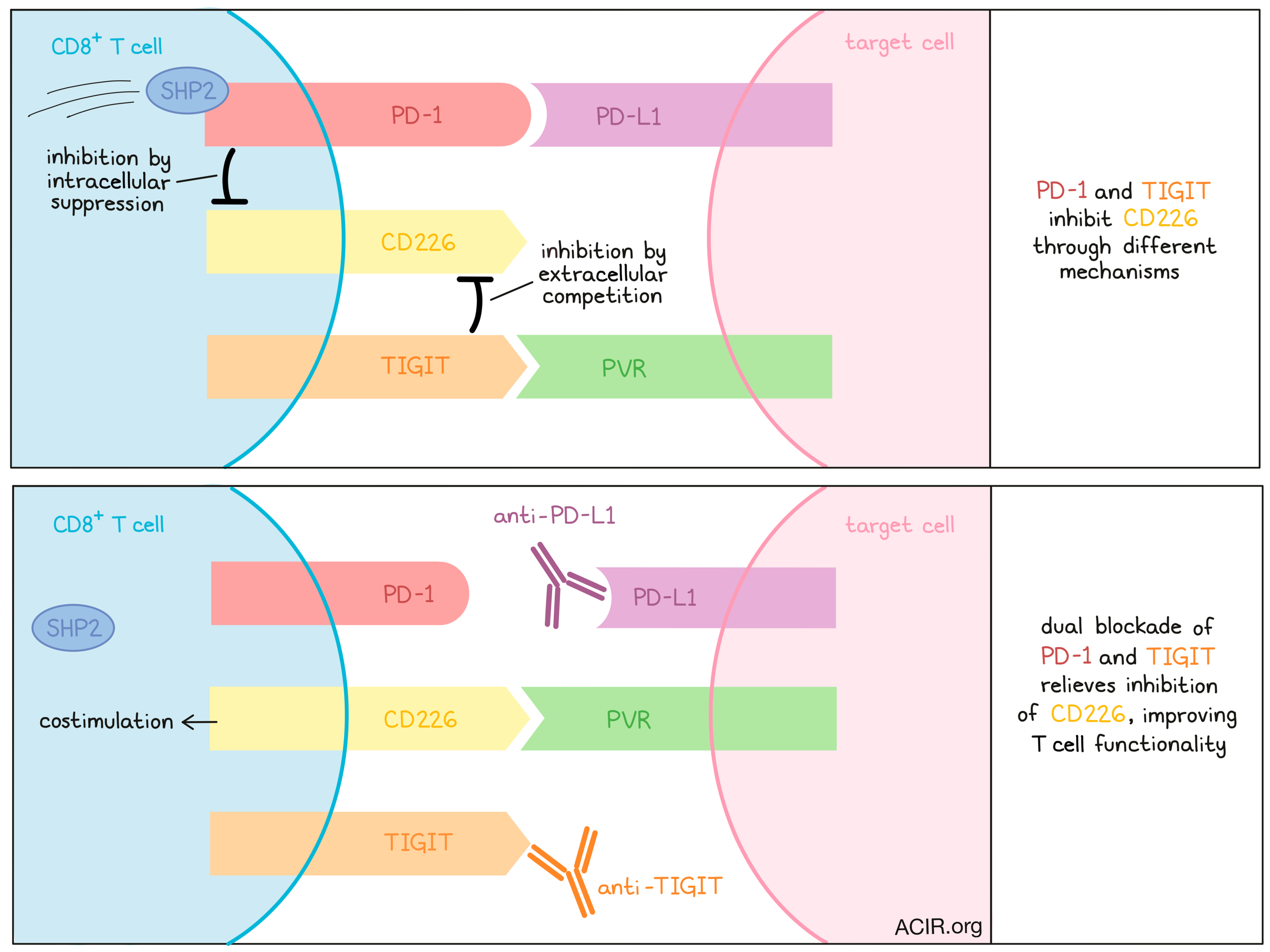
Next, the authors probed more precisely how PD-1, TIGIT, and CD226 interact on T cells. They established a co-culture system between CD226+ Jurkat T cells, which could be further transduced with PD-1 or TIGIT, and antigen-expressing Raji lymphoma cells expressing PD-L1, PVR (TIGIT ligand), or both. CD226 phosphorylation was used as a readout of CD226 activity. Interestingly, the co-expression of either PD-1 or TIGIT on CD226+ Jurkat cells decreased CD226 phosphorylation, indicating that each molecule can inhibit CD226, and this inhibition was maximized when both were expressed. CD226 phosphorylation could be restored by antibody blockade of either PD-1 or TIGIT, but most effectively with dual blockade, altogether demonstrating that PD-1 and TIGIT both inhibit CD226 activity in an additive manner.
Investigating which portion of these receptors mediated CD226 inhibition, the authors found that the intracellular domain (ICD) of PD-1 was required to block CD226 phosphorylation, as a mutant PD-1 lacking the ICD did not induce this effect. However, this was not the case for TIGIT, which could still inhibit CD226 activity without its ICD. A FRET-based assay found that the PD-1 ICD recruited phosphatases Shp2 and Shp1, among other T cell signaling proteins, while the TIGIT ICD did not, potentially indicating that the recruitment of these phosphatases (or associated molecules) was responsible for the dispensability of the TIGIT ICD in CD226 inhibition.
Because TIGIT’s ICD was not required for CD226 inhibition, the authors considered how the extracellular domain of TIGIT might be responsible. They hypothesized that TIGIT could simply outcompete CD226 on the cell surface for its ligand PVR. Testing this theory, they co-cultured CD226+TIGIT+ Jurkat cells with Raji cells expressing PVR at either a high or low density. Theoretically, if PVR ligand availability was the limiting factor for CD226 activation, CD226 phosphorylation should be reduced more strongly in the low-PVR case. Indeed, the researchers observed reduced CD226 phosphorylation at the lower PVR concentration, suggesting that the TIGIT extracellular domain outcompetes CD226 for its ligand.
Finally, the authors used confocal microscopy to visualize the Jurkat-Raji cell interface. CD226 localized to the interface, but less so when TIGIT was co-expressed (which was also enriched at the interface). CD226 polarization could be restored by TIGIT antibody blockade, and as previously observed, only the TIGIT extracellular domain was required for CD226 interference.
In summary, the authors found that both PD-1 and TIGIT restrict CD226 costimulation on CD8+ T cells, but through different mechanisms: PD-1 by recruiting inhibitory phosphatases to its intracellular domain, and TIGIT by extracellular competition for CD226 ligands. Coupled with phenotypic analysis of CD8+ T cells coordinately expressing both CD226 and CD28, these findings shine light on a novel mechanistic underpinning for this clinically encouraging combination ICB therapy. Additional exploration of these mechanisms may support the further development of effective, non-redundant combination immunotherapy strategies for cancer treatment.
Write-up by Alex Najibi, image by Lauren Hitchings
Meet the researcher
This week, senior author Ira Mellman answered our questions.

What prompted you to tackle this research question?
The rationale really goes back to the early days of PD-1/PD-L1. The field began with the understandable assumption that checkpoint inhibitors acted by reversing T cell exhaustion in the tumor. Expecting this was the case, we were surprised in our early mechanistic and clinical biomarker studies of atezolizumab that this appeared not to be the case, rather that PD-L1/PD-1 blockade had a marked effect on T cell function much earlier in the pathway, namely at or around the so-called Tscm compartment, which was activated as a consequence of dendritic cell stimulation. TIGIT, along with PD-1, appeared to be present on these early T cells, or at least T cells poised for activation and proliferation. So, TIGIT’s mechanism also became of interest. In addition, given pre-clinical and now clinical data attesting to the positive benefit of combining PD-L1/PD-1 blockade with anti-TIGIT, we became interested in understanding how the two pathways interacted and why combinations were generally more effective than PD-L1/PD-1 inhibition alone. Further work on TIGIT is indicating also that there remains a lot more to learn about how this molecule works, and the significance of signaling via CD226 in various cell types.
What was the most surprising finding of this study for you?
It was unexpected to find that there was such a close mechanistic relationship between the PD-L1/PD-1 and TIGIT pathways. For a variety of reasons, this appears to “make sense”, especially given human genetics indicating that T cell immunity is not wholly dependent on CD28 costimulation, implicating CD226 as playing a more important role than we previously thought.
What was the coolest thing you’ve learned (about) recently outside of work?
Learning how important Draymond Green is to the success of the San Francisco Warriors. So much else learned of late about the behavior of our fellow human beings has been profoundly depressing.



