Pancreatic ductal adenocarcinoma (PDAC) has a poor prognosis and is largely refractory to current clinical immunotherapies. Freed-Pastor et al. investigated the immune evasion mechanisms in this cancer type by assessing neoantigen load, neoantigen-specific T cells, and T cell characteristics, revealing a novel axis of inhibition. Their results were recently published in Cancer Cell.
Since there has been debate about the level of neoantigens that PDAC harbors, Freed-Pastor et al. assessed the broader neoepitope landscape by developing a neoepitope prediction pipeline that included HLA allele typing, mutation calling, variant effect prediction, and variants derived from missense, frameshift, and inframe insertion/deletion mutations. They also used peptide:MHC class I binding predictions that considered relative binding of the wild-type sequence. Confirming earlier work, most early-stage tumors in TCGA harbored putative neoepitopes, and 73% of patients expressed neoepitopes with a predicted high affinity for MHC-I. When assessing those neoepitopes with a higher affinity for MHC-I than wild-type peptide, or for which no corresponding wild-type existed, 81% of patients harbored these “non-binding-to-binding” neoepitopes. Additionally, all advanced/metastatic tumor samples harbored predicted neoepitopes, of which 87% contained high-affinity epitopes and 98% contained “non-binding-to-binding” neoepitopes.
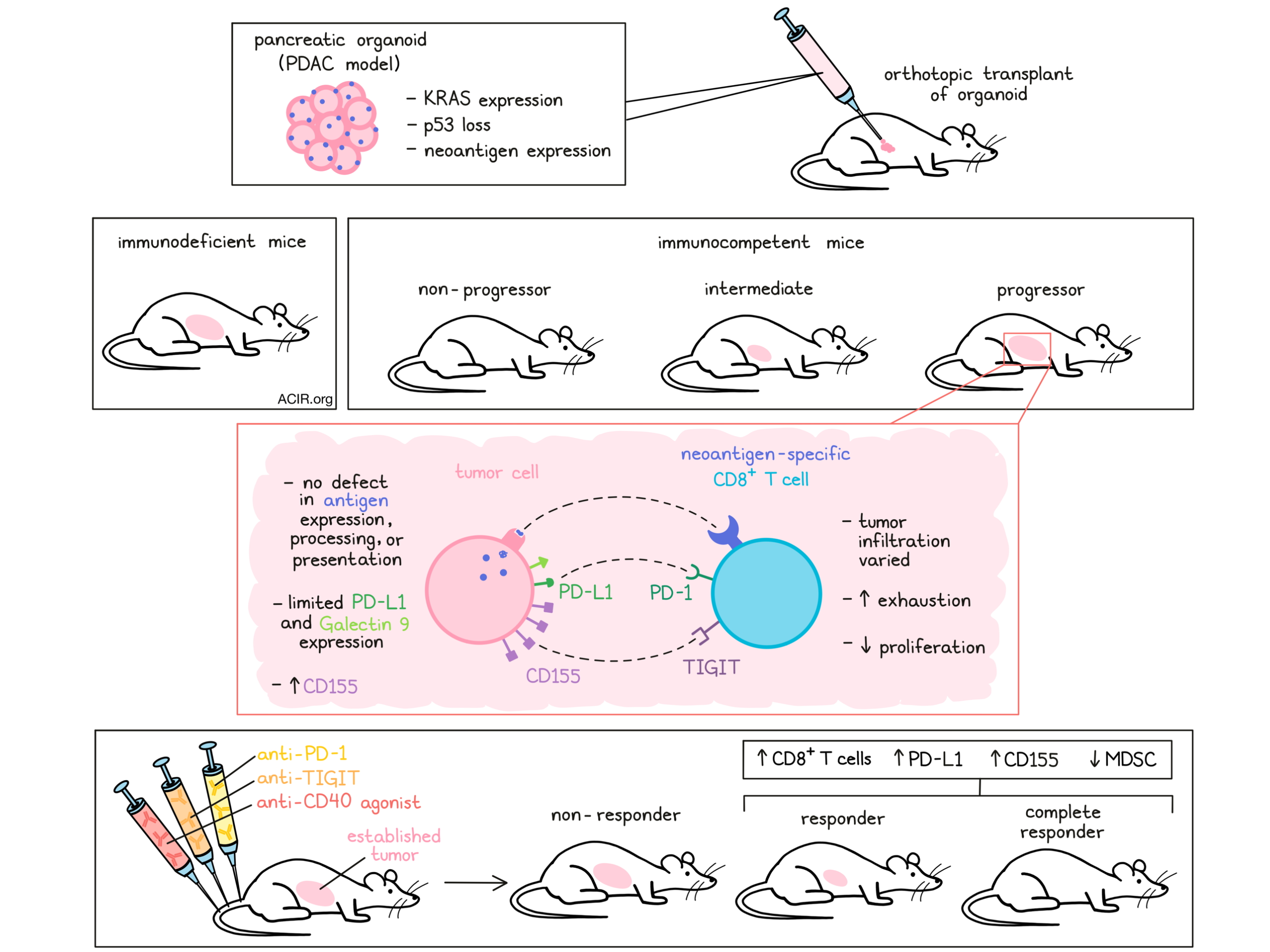
To evaluate the molecular and cellular mechanisms of immune evasion in this cancer type, Freed-Pastor et al. used CRISPR/Cas9-assisted homology-directed repair to generate knock-ins of defined neoantigens, including ovalbumin (OVA) and endogenously arising MHC class I-restricted neoantigens. Using this technology, “genetically defined” pancreatic organoids were cultured from tumors from animals that expressed mutated KRAS and had a loss of p53, and had stable and uniform neoantigen expression. The organoids were successfully orthotopically transplanted into the pancreas of CD8+ T cell-depleted or Rag2-/- immunodeficient mice. In immunocompetent mice on the other hand, tumor cells were either rejected (non-progressor), were present but did not develop macroscopic tumors (intermediate), or grew into macroscopic tumors (progressor). Even a proportion of tumors expressing the highly immunogenic neoantigen OVA evaded immune destruction while maintaining antigen expression. This progressor subset was further studied to assess the mechanisms behind this immune evasion.
These immune-evasive tumors did have CD8+ TIL, but there was inter- and intra-tumoral heterogeneity in infiltration patterns, similar to human PDAC. The researchers hypothesized that immune-evasive tumors had either acquired defects in antigen processing and presentation, or TIL had become dysfunctional. To investigate this, the researchers re-isolated organoids from immune-evasive and immunodeficient mice. There were no differences in neoantigen, MHC-I, or MHC-II expression. Furthermore, an organoid and antigenically stimulated OT-I CD8+ T cell co-culture system showed no loss of killing in the immune-evasive tumors. Therefore, loss of antigen expression or antigen processing and presentation was unlikely to cause this immune evasion.
The phenotype of neoantigen-specific T cells was determined to validate the hypothesis that dysfunctional T cells were responsible for the immune evasion. Even though there was no difference in the abundance of neoantigen-specific cells in progressors as compared to non-progressors, there was a decrease in proliferative capacity over time in the immune-evasive tumors. Furthermore, these T cells also expressed two or more co-inhibitory receptors, suggestive of a dysfunctional phenotype.
Neoantigen-specific TIL were subjected to single-cell RNA-seq and clustered into four main clusters, of which the largest cluster was enriched for genes associated with T cell exhaustion and a chronic effector signature. The smaller clusters were enriched for naive and memory markers, and inhibitory receptors marking CD8+ Tregs. TIGIT and PD-1 co-expression best differentiated the neoantigen-specific from the non-specific subsets.
To assess whether similar patterns could be found in human PDAC, the researchers isolated intratumoral CD8+ T cells from resected tumors and assessed their phenotype. The majority of CD8+ TIL were CD45RO+, indicating antigen experience, and a subset co-expressed multiple co-inhibitory receptors. The dysfunctional/exhausted phenotypes, as found in the murine models, were also detected in a previously published human scRNAseq dataset.
The researchers then assessed the expression of the ligands of multiple inhibitory receptors. PD-L1 and Galectin 9 (ligands of PD-1 and TIM-3, respectively) expression was limited in PDAC tumors, but CD155 (aka PVR and the ligand of TIGIT) expression was present on both murine and human tumors. The expression of CD155 was significantly higher in human tumor samples than in healthy pancreas; however, approximately 20% of tumors lacked expression. Additionally, immune-evasive tumor organoids and organoids obtained from immunodeficient mice expressed CD155. Assessing various isogenic mice, it was shown that CD155 expression significantly increased when there was concomitant oncogenic KRAS expression and p53 loss, data that were confirmed in PDAC samples from TCGA. Going back to the neoepitope data, tumors with a high predicted neoepitope burden and those with more “non-binding-to-binding” neoepitopes also showed elevated CD155 expression.
Based on these results, the researchers hypothesized that a combination of CD40 agonism – a potent stimulator of APC–T cell interactions that can overcome the need for CD4+ T cell help – and checkpoint blockade might aid in overcoming T cell dysfunction in PDAC. Therefore, immune-competent mice with established orthotopic organoids were treated with mono- or dual PD-1 and TIGIT blockade, with or without CD40 agonism. Neither mono nor dual checkpoint blockade resulted in tumor responses. However, combining both TIGIT and PD-1 blockade with the CD40 agonist resulted in 23% complete responses. Responders had higher levels of CD8+ TIL infiltration, as well as increased PD-L1 and CD155 expression in the stroma. In addition to an increase in CD8+ T cells, responders also had a decrease in immunosuppressive myeloid subsets. In non-responders, myeloid-derived suppressor cells were found in areas without T cells.
The researchers then used a genetically tractable autochthonous and orthotopic (via retrograde pancreatic duct delivery of the CRE-containing lentiviral vector) mouse model of neoantigen-expressing PDAC in immunocompetent and immunodeficient mice. Tumors grew in the immunodeficient mice, but only in a subset of the immunocompetent mice, and in those, the neoantigen expression was downregulated, suggestive of immune editing. When CD155 was forcibly upregulated in this model, there was an increase in the proportion of mice with immune evasion. Similarly, when mice were treated with a TIGIT agonist, there was an increase in immune evasion.
The mechanistic and translational research presented here shows that the CD155/TIGIT axis may play a key role in immune evasion in PDAC, and that incorporating TIGIT axis blockade into combination strategies may aid in improving therapeutic efficacy in patients.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author William Freed-Pastor and lead author Tyler Jacks answered our questions.

What prompted you to tackle this research question?
WFP: Pancreas cancer has historically been considered poorly immunogenic, due to its intermediate mutational burden and relative scarcity of infiltrating T cells. However, this paradigm has been profoundly challenged over the past few years, as recent studies have revealed that the immune landscape in human PDAC is significantly more heterogeneous than previously appreciated. Multiple groups have now identified predicted neoepitopes in a sizable subset of patients with pancreatic cancer and, in fact, multiple groups have now been able to isolate neoantigen-specific T cells from pancreatic cancer patients, providing strong evidence that a subset of human pancreatic cancers harbor neoantigens capable of evoking an endogenous antitumor immune response. This prompted us to develop preclinical models for this subset of patients to better understand how these tumors successfully hide from the immune system.
TJ: For some years, our laboratory has been interested in developing improved pre-clinical models for studying the interactions between a developing tumor and the immune system. This effort has centered on our lung models in the past. Here we describe our first efforts in developing so-called immunogenic models of pancreas cancer. In this case, the need is especially great, given the challenges associated with pancreas cancer treatment and the generally refractory nature of the disease to immunotherapy. Our work, which was led by Will Freed-Pastor and Laurens Lambert, provides a set of pre-clinical platforms to study these questions systematically, but also highlights the role of one immune-modulatory pathway that has received relatively less attention previously: the CD155/TIGIT pathway. This study nicely addresses two of our goals: (1) learn about the complex interactions involving the tumor immune microenvironment and (2) provide the foundation for further investigation in the clinic.
What was the most surprising finding of this study for you?
WFP: One of the most surprising findings was that a sizable subset of pancreas cancers in our neoantigen-expressing organoid-based preclinical models acquired the ability to evade immune surveillance, despite continued expression of a highly immunogenic antigen (SIINFEKL). Prior reports, using neoantigen-expressing two-dimensional (2D) monolayer cell lines, had observed, almost exclusively, tumor rejection using a similar approach. This suggested that preclinical models that better recapitulate the prolonged tumor kinetics and histopathology of human pancreatic cancer could uncover a propensity for immune evasion previously overlooked using monolayer cell lines, and furthermore could be used to query the mechanisms by which these tumors evade immune surveillance.
TJ: I was surprised by several things, in fact, but two findings stand out. On the one hand, we were able to generate a significant fraction of pancreas cancers that developed despite the expression of one or another strong neoantigen. We have seen this before in other settings, including the lung, but I was prepared for this system to result in complete tumor rejection. In fact, about 50% of the tumors in one of our model systems yielded progressing, antigen-expressing tumors. This result was critical for allowing us to investigate the nature of immune suppression in the model, as well as to be able to test various therapeutic regimens. The second surprise was just how well the triple combination therapy worked. The results from the pre-clinical trials are really quite striking and have provided the impetus for planned clinical studies.
What was the coolest thing you’ve learned (about) recently outside of work?
WFP: A few weeks ago, we took our son (11mos) and daughter (2.5yrs) for a day on the town around Boston. We all had a fantastic day and one of the highlights was when the kids got to ride a carousel downtown. As the ride was ending, my daughter nearly had a fit because she didn’t want it to stop. I helped her off and then sat her down and tried to explain to her that it is silly to get upset when something good stops, instead we need to be happy that we got to do it at all. It only took about 10 minutes before it hit me how much I still need to learn that same lesson. It is so easy to see the world today with eyes of frustration and despair, but this life is so beautiful. What response can we ever really have except one of pure gratitude.
TJ: How can we not talk about COVID in response to that question? I have been impressed by the feelings of joy people (including me!) are expressing as life begins to return to normal. Needless to say, we are not out of the woods here, and there is a strong need for continued vigilance, but it is refreshing to start to see people gathering and smiling and really enjoying life again. When I walk my golden retriever Leo around the streets of Boston, I am heartened to see so many smiling faces again.



