Recombinant human interleukin-2 (rhIL-2, aldesleukin) was approved decades ago for the treatment of cancer, and it induces long-lasting, sometimes complete responses in a subset of patients with metastatic melanoma or metastatic renal cell carcinoma. However, due to the risk of capillary leak syndrome and associated toxicities, the use of rhIL-2 treatment has been limited to patients with normal heart and lung function. Given the central role of IL-2 in T cell biology and the desire to improve the efficacy and safety of IL-2-based immunotherapy, Lopes et al. designed and thoroughly tested an IL-2-containing fusion protein. The promising results were recently published in the Journal for ImmunoTherapy of Cancer.
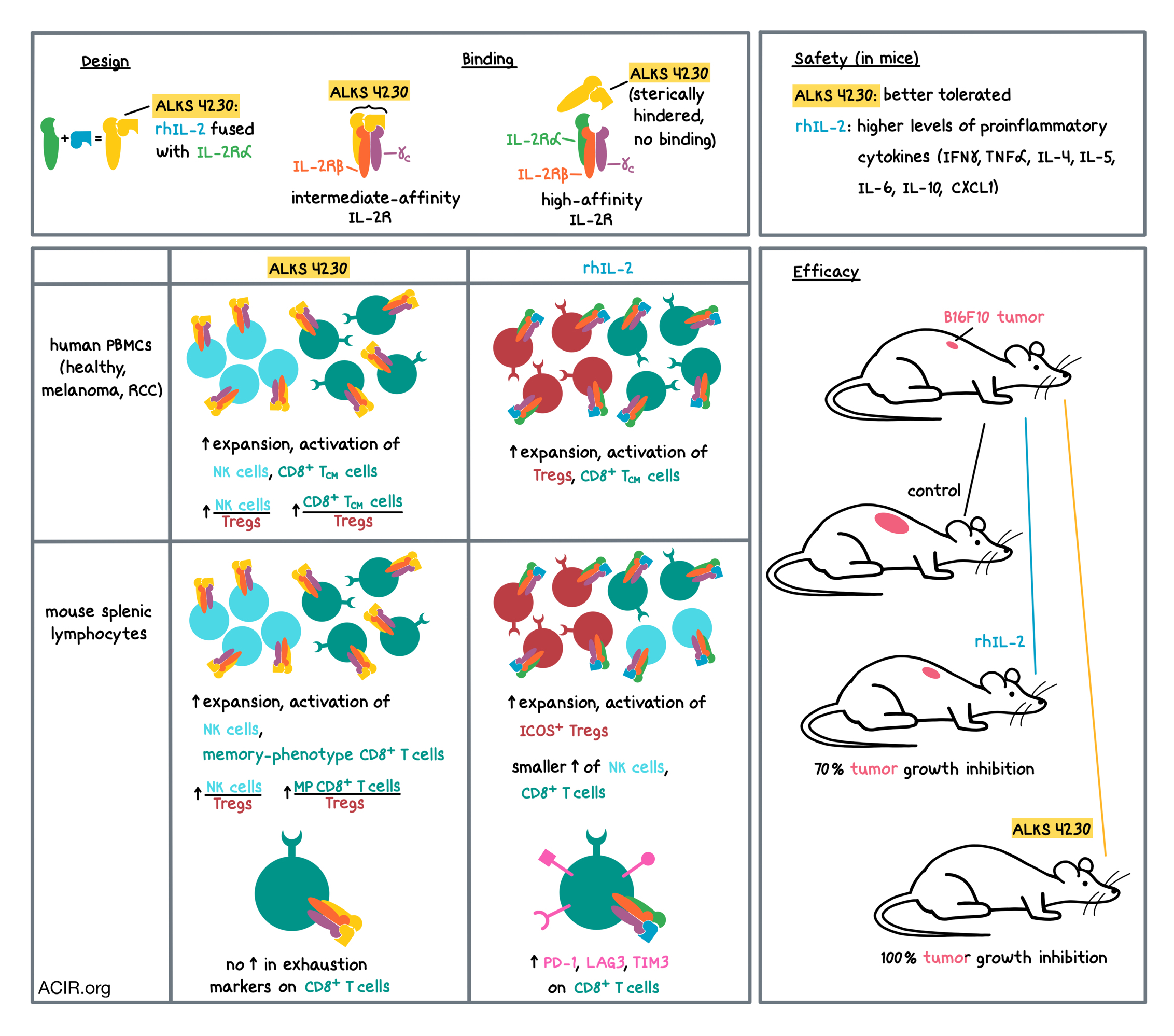
To design a safer and more efficacious IL-2-based treatment, the researchers took into account the different ways that IL-2 signals in various cell populations. At low concentrations, IL-2 signals through the high-affinity IL-2 receptor (IL-2R), which consists of IL-2Rα, IL-2Rβ, and the common gamma chain (γc). Higher concentrations of IL-2 are required to signal through the intermediate-affinity IL-2R, which consists of IL-2Rβ and γc. IL-2Rα is constitutively expressed on Tregs, and inducibly expressed upon activation on NK cells and effector T cells, while IL-2Rβ and γc are constitutively expressed on NK cells and memory CD8+ T cells. Thus, in addition to the safety concern of capillary leak syndrome, rhIL-2 may also be undermining its antitumor effects by activating and expanding immunosuppressive Tregs.
Keeping the expression patterns of IL-2 receptors in mind, Lopes et al. designed ALKS 4230, a recombinant human IL-2 covalently fused with the extracellular domain of IL-2Rα. Using a structure-based design tool called circular permutation, the N- and C-termini of rhIL-2 were joined with a small linker, and the circular chain was broken to create a new C-terminus to which the extracellular domain of the IL-2Rα could be attached. The rhIL-2 portion of the final fusion protein was still able to interact with IL-2Rβ and γc, while the IL-2Rα portion of the fusion protein sterically hindered the rhIL-2 portion from interacting with the endogenous IL-2Rα. Thus, the researchers hypothesized that ALKS 4230 would signal through the intermediate-affinity, but not the high-affinity, IL-2R, and preferentially activate effector lymphocytes over Tregs.
The researchers first tested their hypothesis in vitro. Testing in human cell lines revealed that ALKS 4230 and rhIL-2 both activated cells expressing the intermediate-affinity IL-2R to a similar degree, but ALKS 4230 was much less potent than rhIL-2 in activating cells expressing the high-affinity IL-2R. Similar results were observed in PBMCs from healthy human donors and patients with either advanced melanoma or advanced renal cell carcinoma. In PBMCs cultured with anti-CD3 and either ALKS 4230 or rhIL-2, ALKS 4230 expanded and activated more NK cells, rhIL-2 expanded and activated more Tregs, and both proteins expanded central memory CD8+ T cells to a similar extent. The ratios of NK cells to Tregs and central memory CD8+ T cells to Tregs were both higher with ALKS 4230 than with rhIL-2.
The researchers confirmed in vitro that the effects of ALKS 4230 and rhIL-2 on mouse lymphocytes recapitulated the effects on human lymphocytes, supporting the use of mouse models in analyzing in vivo responses to treatment. Analysis of splenic lymphocytes from mice treated with ALKS 4230 or rhIL-2 showed that, unlike rhIL-2, ALKS 4230 did not increase Tregs. rhIL-2 also increased the proportion of Tregs that were ICOS+, which survive longer and are more immunosuppressive than ICOS- Tregs. ALKS 4230 preferentially increased the populations of memory-phenotype CD8+ T cells and NK cells to a much greater extent than rhIL-2 treatment. Similar to human in vitro data, ALKS 4230 increased the ratios of NK cells and memory-phenotype CD8+ T cells to Tregs, while rhIL-2 did not. The ratio of conventional CD4+ T cells to Tregs was also higher with ALKS 4230 (despite a decrease in conventional CD4+ T cells). rhIL-2 significantly increased the portion of memory-phenotype CD8+ T cells that expressed exhaustion markers PD-1, LAG3, or TIM3, while ALKS 4230 did not. ALKS 4230 also significantly increased the number of the highly mature and cytolytic CD11b+CD27- NK cells compared to rhIL-2 treatment.
The favorable changes in lymphocyte subsets observed with ALKS 4230 came with better tolerability, as assessed by levels of pulmonary edema. The researchers also found that rhIL-2 induced much higher levels of proinflammatory cytokines, including IFNγ, TNFα, IL-4, IL-5, IL-6, CXCL1, and IL-10, compared with ALKS 4230, potentially explaining the more favorable tolerability profile of ALKS 4230 over rhIL-2.
Finally, the researchers tested the two treatments in tumor-bearing mice. Mice with B16F10 melanoma (which colonized the lung) were subcutaneously injected with either rhIL-2 or ALKS 4230 at various doses. The best performing dose of rhIL-2 resulted in 70% tumor growth inhibition compared to control, while the best performing dose of ALKS 4230 led to 100% tumor growth inhibition. ALKS 4230 performed similarly, in a dose-dependent manner, whether administered subcutaneously or intravenously. Testing of different dosing frequencies (varying from daily to once weekly, with doses selected to achieve similar exposure to the drug) of IV or subcutaneous administration of ALKS 4230 revealed no differences in tumor growth inhibition, providing basis for flexibility in clinical trial design.
Overall, in mice, ALKS 4230 showed greater expansion of CD8+ T cells and NK cells, lower expansion of Tregs, lower systemic induction of proinflammatory cytokines and pulmonary edema, and greater antitumor efficacy that was achieved using various dosing schedules and administration routes, suggesting a potentially more favorable risk-benefit profile in the clinic compared with rhIL-2. Based on these preclinical results, several clinical trials have been initiated to evaluate ALKS 4230 for the treatment of patients with advanced cancers.
by Anna Scherer
Meet the researcher
This week, lead author Heather Losey and first author Jared Lopes answered our questions.

What prompted you to tackle this research question?
HL: The primary goal underlying the design of investigational ALKS 4230 was to leverage and expand on the proven antitumor effects of existing IL-2 therapy, while mitigating certain limitations. High-dose IL-2 has demonstrated significant anti-cancer efficacy, however its use in cancer treatment has been limited by its toxicity profile. Therefore, we wanted to explore whether we could apply our protein engineering capabilities to fuse IL-2 and IL-2 receptor α, to create a novel fusion protein, ALKS 4230 that may mitigate the toxicity associated with high-dose IL-2 treatment. We hypothesized that the presence of IL-2 receptor α, stably incorporated as part of the ALKS 4230 molecule, could sterically occlude binding to the high-affinity IL-2 receptor. The high-affinity receptor is expressed primarily on regulatory T cells that can dampen the antitumor immune response, and is also expressed on vascular endothelial cells, thought to mediate side effects of high-dose IL-2 such as vascular leak syndrome.
Personally, I have always been motivated to research new cancer treatments, as my family has been touched by this terrible disease. That, coupled with my background in protein structure and function, prompted me to take up this work.
JL: I’ve had a long-standing interest in understanding immune tolerance, in particular in understanding how regulatory T cells develop, how they function, and how they regulate immune responses through IL-2 yet are dependent on IL-2 for survival and function. Previous work in the field has shown that targeting IL-2 receptors using complexes of IL-2 and anti-IL-2 antibodies could alter the balance of immunosuppressive regulatory T cells and immune effector cells that are expanded in response to IL-2 treatment. Therefore, we set out to design a molecule that could selectively activate the intermediate-affinity IL-2 receptor to specifically stimulate immune effector cells. We theorized that such a selective activation would drive strong antitumor immune responses without directly evoking responses by regulatory T cells. In the work that followed and is now described in this article, we demonstrated this approach through rigorous pre-clinical characterization of our novel investigational molecule ALKS 4230.
What were the most surprising findings of this study for you?
HL: The crystal structure of investigational ALKS 4230 confirmed that ALKS 4230 mimicked the known structure of IL-2 bound to IL-2Rα, and as such retained full potency in activating mouse CD8+ T cells and NK cells, which have tumor-killing capacity. However, ALKS 4230 was ~1000 times less potent than IL-2 in activating regulatory T cells, which are known to suppress cancer-fighting immune mechanisms. The data support that the protein engineering approach we used can yield a molecule with intrinsic selectivity for the intermediate-affinity IL-2 receptor. Based on these data, investigational ALKS 4230 has progressed into multiple clinical studies, and it is really exciting for me to be involved in leading a program that has the potential to help advanced cancer patients.
JL: The selective expansion of subsets of mouse CD8+ T cells and NK cells over regulatory T cells was striking. By contrast, IL-2 treatment preferentially induced the activation and expansion of regulatory T cells, as demonstrated by increased numbers of cells and increased expression of ICOS, GITR, and CD39. As hypothesized, these differences in the immunological response to treatment correlated with significantly enhanced antitumor efficacy of investigational ALKS 4230 relative to IL-2 in the murine mouse model of lung tumor colonization.
What was the coolest thing you’ve learned (about) recently outside of work?
HL: We have all been living in the midst of the COVID-19 pandemic, and it has definitely affected the way I live and work. It is great to see the resilience around us, including physicians and scientists, and hopefully we continue to collaborate to address the challenges of treating and preventing SARS-CoV-2 infection. In addition to being a scientist, I’m also a cellist in the Boston Philharmonic Orchestra. Since the orchestra had to cancel the rest of this season, I really miss the opportunity to rehearse and perform with my fellow musicians. Despite these challenges, it is amazing to see the creativity of musicians and artists coming together in a virtual world, though we are all craving in-person connections.
JL: Recent events have taught me just how passionate, committed, and brave are the people that work to provide care in hospital emergency rooms. This passion energizes me as a scientist, as the whole world is now depending on the scientific and medical communities to develop novel vaccines and treatments to address the COVID-19 pandemic. I think these events will also encourage young students to become scientists and medical professionals to take on future challenges.



