Interleukin 12 (IL-12) is a heterodimeric cytokine with potent antitumor efficacy based on its abilities to stimulate the proliferation and cytotoxicity of activated T and NK cells, induce IFNγ and other cytotoxic enzymes and cytokines, and promote differentiation into memory phenotypes. Its actual efficacy as an immunotherapy, however, is limited by its short half-life and dose-related toxicity. In an effort to harness the positive effects of IL-12 and bypass the negatives, Jung et al. constructed an Fc-fused version of monovalent IL-12 and observed remarkably improved effects, which were reported in Oncoimmunology.
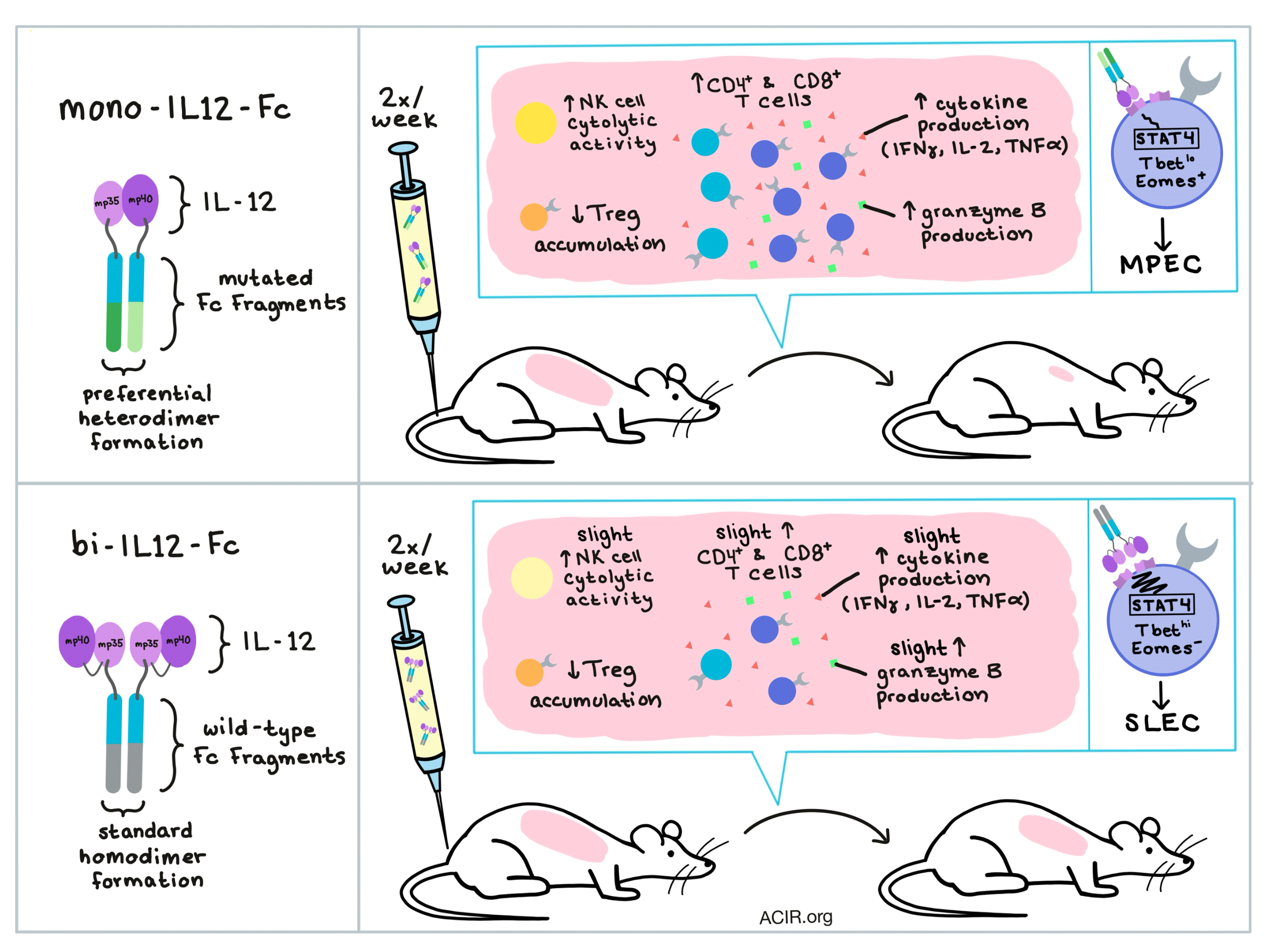
To generate monovalent Ig Fc-fused murine IL-12 (mono-mIL12-Fc), each of the two subunits of IL-12 (p40 and p35) were fused separately to the N-terminus of one of the two respective engineered Fc chains; these Fc chains were mutated such that only heterodimeric Fc would form, ensuring that each dimer would include both subunits of IL-12. As a control, the researchers also generated Fc-fused bivalent IL-12 (bi-mIL12-Fc), which consisted of linked IL-12 subunits attached to a wild-type Fc chain, forming homodimers.
Both mono-mIL12-Fc and bi-mIL12-Fc had longer half-lives compared to recombinant IL-12, and twice-weekly injections of either mono-mIL12-Fc or bi-mIL12-Fc proteins completely cured small CT26-HER2/neu tumors in mice. Jung et al. then went on to test different concentrations of their immunocytokines twice weekly in larger, more challenging tumors (CT26-HER2/neu, CT26, and B16F10), where mono-mIL12-Fc consistently outperformed bi-mIL-12-Fc, eliciting robust tumor regression and durable cures, even at fairly low doses. At the optimal dosing regimen tested, 73% of mice treated with mono-mIL12-Fc experienced tumor-free survival. Even a single dose of mono-mIL12-Fc at a higher concentration substantially impeded tumor growth in almost 50% of tumor-bearing mice. Bi-mIL12-Fc had antitumor efficacy as well, but was consistently inferior. All treatments were well tolerated, and cured mice rejected rechallenge.
To understand the observed antitumor effects, the researchers explored the underlying cellular mechanisms 27-35 days after tumor inoculation and IL-12 treatment, and found that mono-mIL12-Fc significantly increased the numbers of CD4+ and CD8+ T cells in both spleens and tumors and reduced accumulation of Treg cells in the tumor. A closer look at the tumor microenvironment showed that mono-mIL12-Fc treatment increased IFNγ production in tumor-bearing mice, and that tumor-infiltrating T cells (especially CD8+ T cells) expressed elevated levels of cytokines including IFNγ, IL2, and TNFα; in addition, CD8+ T cells showed increased levels of granzyme B. Further, splenic T cells treated with mono-mIL12-Fc were more cytotoxic when tested against tumor target cell lines in vitro. NK cells also showed increased cytotoxicity, suggesting a partial contribution to tumor control.
Jung et al. next explored whether mono-mIL12-Fc and bi-mIL12-Fc differentially affected the generation of memory CD8+ T cells, and thus analyzed T cell subsets in the spleen after therapy, classifying CD8+ T cells as either effector, effector memory (TEM), or central memory (TCM), based on their expression levels of CD62 and IL7R. While low-dose bi-mIL-Fc treatments slightly increased populations of TEM and TCM cells, mono-mIL12-Fc significantly increased both populations in a dose-dependent fashion.
The researchers next analyzed the induction of a memory phenotype by evaluating the proportions of short-lived effector cells (SLECs) versus memory precursor effector cells (MPECs), which can be differentiated by expression levels of the terminal differentiation marker KLRG1 or the memory marker IL-7R. They found that mono-mIL12-Fc moderately transduced IL-12 signaling to cause weak STAT4 activation in CD8+ T cells, yielding low T-bet expression and more incidents of Eomes expression, thus programming effector CD8+ T cells to differentiate into MPECs. Bi-mIL12-Fc, on the other hand, potently transduced IL-12 signaling, triggering strong STAT4 activation, high T-bet expression, and prevention of Eomes expression, directing CD8+ T cells to differentiate into SLECs.
To better understand how these findings might translate to a clinical setting, Jung et al. developed human versions of both mono- and bi-mIL12-Fc. Both fusion proteins potently induced proliferation of activated human PBMCs in vitro. As in mice, mono-hIL12-Fc also led to reduced expression of pSTAT4 and T-bet compared to bi-hIL12-Fc in stimulated human CD8+ T cells.This indicates that, like its murine analog, mono-hIL12-Fc could induce CD8+ T cell differentiation into MPECs for a durable memory T cell response in humans, supporting clinical evaluation.
by Lauren Hitchings



