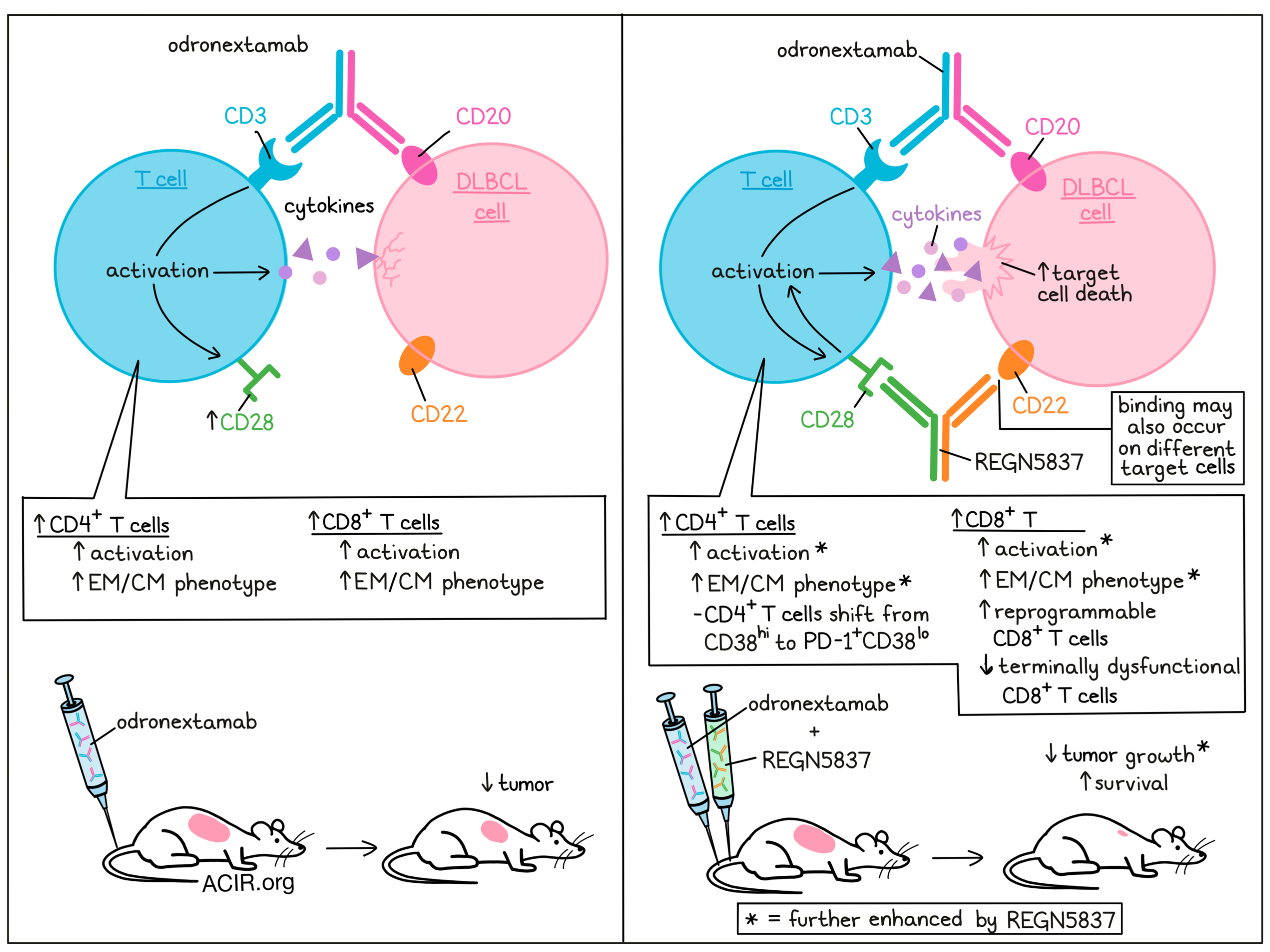
Patients with diffuse large B cell lymphoma (DLBCL) are given rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone as a first-line treatment, and while this therapy is quite effective, 30-40% of patients are refractory or relapse, and face a poor prognosis with second- and third-line treatments. Although some bispecific T cell engagers that provide “signal 1” via CD3 costimulation have shown promise in this setting, Wei et al. noticed room for improvement, and sought to explore the addition of bispecifics that provide “signal 2” through CD28 costimulation. In a recent study, Wei and colleagues investigated the addition of REGN5837 (a CD22xCD28 bispecific) to odronextamab (a CD20xCD3) bispecific. Their results were recently published in Science Translational Medicine.
REGN5837 is a hinge-stabilized human IgG4-based antibody targeting CD22 – a transmembrane protein found on normal B cells and malignant B cells – and CD28 – a costimulatory molecule that is upregulated on CD3-activated CD8+ and CD4+ T cells, like those induced in DLBCL patient samples following treatment with odronextamab. Unlike CD28’s natural ligands, CD80 and CD86, REGN5837 does not bind to CD28’s inhibitory competitor, CTLA-4, providing a costimulatory advantage.
Using lymphocyte-enriched PBMCs cocultured with WSU-DLCL2 human DLBCL cells (expressing high levels of CD20 and CD22) as target cells, Wei et al. investigated the effects of several different doses of odronextamab alone or in combination with a fixed dose of REGN5837. In this setting, REGN5837 enhanced odronextamab-mediated cytotoxicity and contributed to enhanced activation of CD4+ and CD8+ T cells, as measured by increased proliferation, CD25 expression, and cytokine release. Similar results were observed with NALM6 B-ALL cells and Burkitt’s lymphoma Raji CD80/CD86dko cells. Further, while odronextamab or odronextamab plus REGN5837 was ineffective against CD22-deficient target cells, treatment with the single-agent or combination induced strong T cell-mediated cytotoxicity against both CD22+ and CD22- cells in mixed cocultures of the target cells, even when as few as 20% of cells expressed CD22. The addition of REGN5837 was found to contribute to the enhanced lysis of CD22+ and particularly CD22- cells. Similar results were observed in Toledo DLBCL cells, which express lower levels of CD22 than WSU-DLCL2.
Moving into mouse models, the researchers utilized mice bearing WSU-DLCL2 tumors. In this model, prophylactic treatment with odronextamab induced an initial response, but was insufficient to clear tumors, with all mice eventually succumbing to their tumors. However, the addition of REGN5837 (which induced no effects as a monotherapy) resulted in tumor rejection in 6 out of 7 (86%) mice and significantly prolonged overall survival. In a therapeutic treatment setting, administration of odronextamab 26 days after tumor transplantation led to a significant reduction in tumor mass, which was further enhanced with the addition of REGN5837. Further investigation showed that odronextamab decreased the number and density of tumor cells while increasing the expansion and density of CD4+ and CD8+ T cells, particularly central memory and effector memory CD8+ T cells. The addition of REGN5837 further enhanced these effects, particularly in CD8+ T cells. Similar results were observed in mice bearing Toledo tumors and in a mouse model of systemic B-ALL.
Acknowledging the caveat that the DLBCL tumor cells used in their previous models were mixed with human PBMCs before implantation, Wei et al. implanted WSU-DLCL2 tumors into human immune system (HIS; SIRPAh/hTPOh/mRag2−/−Il2rg−/−) mice engrafted with CD34+ liver cells. In this setting, odronextamab alone was once again insufficient to clear tumors (0% of mice alive at day 64), while combination treatment with REGN5837 suppressed tumor growth and extended survival (80% of mice alive at day 64). Interestingly, REGN5837 monotherapy also induced tumor rejection in this setting, due to the allogeniecity between donor T cells and WSU-DLCL2 tumor cells providing signal 1.
The researchers found that In the periphery, odronextamab monotherapy increased CD8+ T cells and production of serum cytokines while depleting circulating B cells. While REGN5837 monotherapy induced no effects in the periphery, the addition of REGN5837 to odronextamab enhanced expansion of CD4+ and CD8+ T cells and increased cytokine release, suggestive of enhanced T cell activation. In tumors, REGN5837 alone induced some T cell infiltration and T cell-mediated killing of tumor cells. However, odronextamab induced stronger T cell-mediated antitumor effects, which could be further enhanced with the addition of REGN5837. Odronextamab and combination treatment also both drove the expansion of effector memory (EM) CD8+ and CD4+ T cells. Further analysis of T cell populations showed that combination treatment enriched a population of CD38loCD101lo CD8+ T cells with reversible dysfunction, while suppressing a terminally dysfunctional CD8+ T cell population that is highly proliferative, but lacks cytokine production and is associated with tumor progression. Combination treatment also induced a shift in CD4+ T cells from CD38hi to PD-1+CD38lo – a population that has previously been associated with better tumor control due to the maintenance of stemness and effector functions and the prevention of metabolic exhaustion.
In cynomolgus monkeys, Wei et al. showed that odronextamab, REGN5837, and combination treatment were well tolerated, with no early deaths, life-threatening events, adverse test article-related clinical findings, or signs of cytokine release syndrome at any of the tested doses. REGN5837 alone did not deplete B cells or induce peripheral CD4+ or CD8+ T cell expansion or activation in this setting, consistent with the absence of signal 1. Odronextamab, on the other hand, depleted peripheral B cells and induced an initial lymphocyte margination, followed by a trend toward expansion and activation of CD4+ and CD8+ T cells. The addition of REGN5837 significantly expanded and activated peripheral CD8+ and CD4+ T cells, though at a high dose, REGN5837 became less effective. This bell curve may be due to excess antibodies saturating each target site by one-armed binding (dimers) and a failure to form the necessary trimer cross-linking between T cells and their targets. Interestingly, higher doses of odronextamab alone induced higher levels of serum cytokines than the lowest dose of odronextamab with REGN5837, though they induced similar levels of CD4+ and CD8+ T cells.
Overall, Wei et al. show that treatment with a CD3-stimulating bispecific, like odronextamab, can be enhanced with the addition of a CD28-stimulating bispecific, like REGN5837. These bispecifics synergized in vitro and in vivo, mediating curative antitumor responses in preclinical models of DLBCL by enhancing both the quantity and quality of activated T cells, providing support for the clinical investigation of REGN5837 in combination with odronextamab in patients with relapsed/refractory DLBCL.
Image and write-up by Lauren Hitchings
Meet the researcher
This week, lead author Dimitris Skokos answered our questions.
What was the most surprising finding of this study for you?
Odronextamab, an investigational CD20xCD3 bispecific antibody that provides “signal 1” through the activation of the T cell receptor/CD3 complex, has exhibited early, promising activity for patients with highly refractory DLBCL in phase 1 trials. However, not all patients achieve complete responses, and many relapse, thus representing a high unmet medical need. This study represents the first preclinical evidence that our costimulatory bispecific antibody (CD22xCD28) may be able to markedly enhance the antitumor activity of Odronextamab (CD20xCD3) in preclinical NHL models, and that the combination of these two bispecific antibodies may provide a chemotherapy-free approach for the treatment of DLBCL. It is worth noting that this is our third publication, in addition to Skokos et al. STM 2020, & Waite et al. STM 2020, related to the role of costimulatory bispecifics in providing the missing costimulatory “signal 2” and improving the antitumor response to CD3-bispecifics and/or checkpoint inhibitors. Across these three preclinical studies, we were able to reproduce and confirm our findings using different tumor-associated antigens (TAAs), dose regimens, various sophisticated animal disease models, and combination strategies.
What is the outlook?
The idea is to use revolutionizing technology platforms together with innovative science that will hopefully enable us to change the narrative and approach cancer as a disease that can be managed. We have now created an entire class of costimulatory CD28 bispecifics that can be combined with our foundational drug cemiplimAb (a-PD-1) and/or our CD3 bispecific antibodies, allowing us to tailor the immunotherapeutic strategy based on the immunological landscape of the tumor. Currently, we are testing three costimulatory bispecific antibodies (PSMAxCD28, REGN5678; EGFRxCD28, REGN7075; MUC16xCD28, REGN5668) in the clinic in various solid tumors, and we hope to initiate a phase I clinical trial with our CD22xCD28 (REGN5837) in combination with Odronextamab (CD20xCD3) in heme malignancies (DLBLC patients). Recently, we communicated preliminary clinical data showing encouraging dose-dependent antitumor activity of investigational PSMAxCD28 when combined with a standard dose of cemiplimAb, suggesting potential to overcome metastatic castration-resistant prostate cancer (mCRPC), resistance to PD-1 inhibition. Now that our xCD3 and xCD28 bispecific platforms continue to be researched in the clinic, along with our anti-PD-1 and anti-LAG3 checkpoint inhibitors, we are meticulously developing and evaluating future combinations for various tumor types.
What was the coolest thing you’ve learned (about) recently outside of work?
I have always been fascinated by the curiosity, imagination, and spontaneity of kids. During the pandemic, I got the chance to spend a bit more time with my 4-year-old daughter, who actually is trilingual (English, French, and Greek). Witnessing the evolution of her linguistic skills, and her ability to keep learning and improving in various languages while wearing a face mask for over a year in school reminded me of the ability of the human mind to adapt and identify ways to work around various obstacles that come our way.



