
The ACIR team attended the ESMO Targeted Anticancer Therapies Congress 2024 in Paris, France. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Keynote lecture on tumor neoantigens
Yardena Samuels
Bispecifics
Juanita S. Lopez
Shohei Koide
Georg Falck
Antibody-drug conjugates
Takashi Kagari
Giuseppe Curigliano
Barbara Pistilli
Toshio Shimizu
Targeted interleukins
Giuseppe Curigliano
Ignacio Melero
Adoptive cell therapy
Michael O’Dwyer
Ronnie Shapira-Frommer
Evelyn Ullrich
Patrizia Porazzi
Keynote lecture on tumor neoantigens
Keynote lecture: Revisiting the neoantigen approach for cancer immunotherapy- Yardena Samuels (Rehovot, Israel)
Presenting the Keynote lecture for the conference, Yardena Samuels described her journey as a cancer geneticist, examining the impact of specific mutations in both the genesis of cancer and the production of neoantigens that support the immune rejection of the cancer. Revitalization and generation of cancer-specific T cells underlies the activity of checkpoint blockade and other immune modalities, such as cancer vaccines and some bispecific antibody and adoptive cell therapy approaches. Transitioning from her pioneering work with Steven Rosenberg in using genomics to identify patient-specific immunogenic neoantigen mutations, Samuels discussed developing immunopeptidomics to examine the physical presentation of neoantigens on HLA as a more direct approach to truly functional neoantigens involved in tumor rejection. Peptidomics became a key tool in her systematic discovery and immune validation of allele-specific recurrent neoantigens from multiple driver genes across a range of cancers, ultimately leading to a library of validated targets for which vaccine or TCR-directed therapy could be used for a multitude of patients. However, given the multiple filters that impact the ultimate utility of a given mutation and the known responsiveness of patients to checkpoint blockade, even when they have a low mutation burden, Samuels hypothesized that there are many more sources of neoantigens that are not readily identifiable using current genomics approaches – the dark proteome. Work in Samuels’s lab in collaboration with Jennifer Wargo showed that microbial peptides were often presented by melanoma cells, and other research groups have shown that non-canonical translation, often of open reading frames in 5’ and 3’ UTRs, leads to presentation of novel peptides. Samuels next focused on novel peptides produced under conditions of cell stress, such as during exposure of cells to high levels of IFNγ released by T cells, which leads to metabolic changes in tumor cells. One such metabolic effect is the reduction of tryptophan (TRP), leading to uncharged TRP-tRNA, ribosome stalling at TRP codons, and eventual “slippage” at particular sites in the reading frame. This slippage produces novel frame-shifted peptide sequences, which are often immunogenic due to both their novelty as neoantigens and the instability of the aberrant protein, which leads to enhanced degradation and presentation. To induce cancer cells to express such aberrant peptides, Samuels’s recent work has focused on forcing reading frame shifts by deleting an enzyme known to be critical to tRNA reading frame fidelity – the tRNA methyltransferase TYW2. TYW2 modifies a base in the phenylalanine (PHE) tRNA adjacent to the anticodon. Knockout (KO) of TWY2 led to a reduction of the modified base, and immunopeptidomics then confirmed the production of multiple novel peptides in the KO cell lines. Riboseq demonstrated the ribosome stalling at the PHE codon, specifically at the A site of the ribosome, which should lead to +1 slippage and the absence of PHE in the frame-shifted translation product (which aids in the construction of a immunopeptidomics search database). Use of a reporter system with a known “slippery” sequence demonstrated the slippage of the reading frame at the protein level. Peptides identified in the TWY2 KO cell line were immunogenic in vitro, and the KO cell line grew more slowly in immunocompetent mice than in immunodeficient mice, adding a tool to expand the repertoire of targetable recurrent neoantigens.
Bispecifics
Beyond CD3 bispecifics: Activating NK cells, CD40, CD137- Juanita S. Lopez (Sutton, United Kingdom)
As an introduction to the session on bispecific antibodies, Juanita S. Lopez provided a broad overview of the field, describing the substantial progress made since the first bispecific T cell engager, Blinatumomab, was approved in 2014, as well as the new directions that are being explored. T cells are the critical warriors in the immune attack against cancer, but are in a constant evolutionary battle against immune-evading tumors. A key early event in natural T cell recognition of tumors is CD3 pathway stimulation following TCR recognition, and so the first bispecific product concept was using CD3 to bridge a tumor-specific target with an activated T cell, as the six recent (2022-2024) approvals have done. However, using CD3 to redirect T cells can result in toxicity, off-target activation of naive T cells following loss of the re-directing antigen in the tumor, and targeting of T cells that are already highly exhausted. Instead, using other co-stimulatory molecules as the T cell-redirecting component may provide alternate biological advantages. For example, targeting CD137 (4-1BB) may limit activation to antigen-experienced cells in the TME, where CD137 is upregulated, thereby reducing toxicity. It may also enhance proliferation and be helpful in promoting immune memory. Bispecifics targeting CD137 and a variety of tumor surface molecules are currently in clinical development. Multiple other opportunities for optimization are also available, including Fc alterations, maximizing the relative affinity for each targeting arm, better pharmacokinetic properties, cis targeting, and combinations with other immunotherapies – especially checkpoint blockade. Particularly interesting are bispecifics that focus activation within the TME by targeting PD-L1 and CD137. Finally, to move beyond T cell activation is the use of CD16 to target activation of natural killer (NK) cells, with several NK cell engagers targeting multiple different targets currently in clinical development.
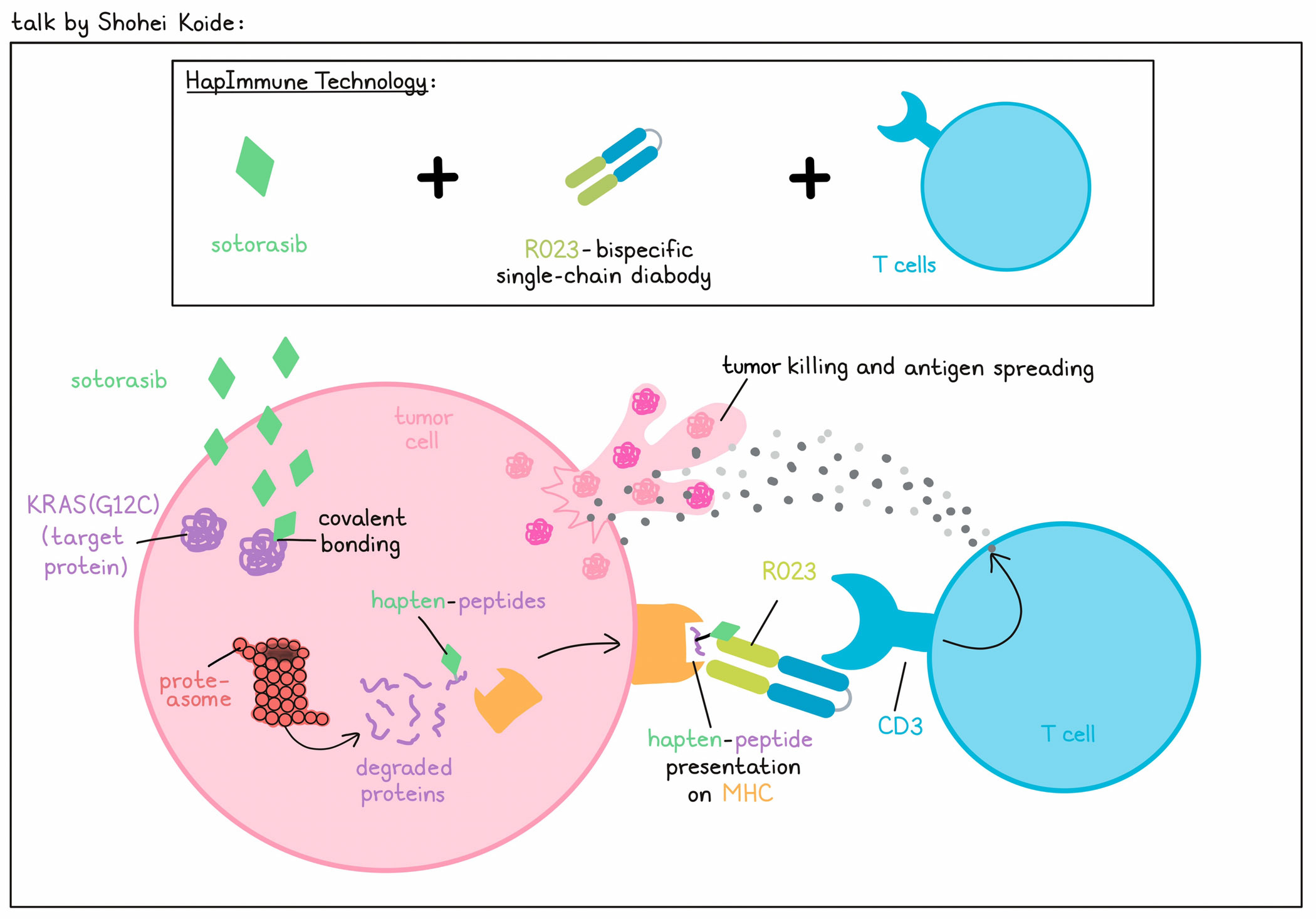
Beyond HLA-restriction: Hapimmune bispecifics- Shohei Koide (New York, United States of America)
Instead of "passively waiting for cancer cells to produce actionable neoantigens", often with small physico-chemical differences from the cognate wild-type molecule, Shohei Koide, together with colleague Benjamin Neel, came up with a novel approach, called HapImmune Technology, to create synthetic cancer-specific antigens and synthetic binders to target them. To target specific proteins and create synthetic antigens, HapImmune Technology utilizes the FDA-approved KRAS(G12C) inhibitor sotorasib, which forms a covalent bond with the mutation-induced cysteine at position 12. Upon proteasomal degradation, peptides with covalently bound sotorasib attached (hapten-peptides) may be presented on MHC. Then, the researchers can use a bispecific antibody that co-targets these drug-peptide/MHC complexes on tumor cells and CD3 on T cells to induce tumor cell killing, antigen spreading, and durable antitumor immunity. To demonstrate this technology, Koide predicted a 9mer and a 10mer KRAS(G12C) peptide to be bound to HLA-A*03 and HLA-A*11, and produced 4 drug-peptide/HLA complex combinations. Screening of a synthetic antibody library via phage display, followed by yeast display, antibody discovery, and maturation, yielded the R023 Fab, which had a high affinity for all four hapten-peptide/HLA complexes. Importantly, antibody binding was not effectively inhibited by the free drug. R023 was then used to create a single-chain diabody (scDb) T cell engager. In a cell-killing assay, the sotorasib-resistant KRAS(G12C) HLA-A*03-positive cell line H2122 was first treated with sotorasib to allow for the formation of drug-peptide/HLA complexes before the addition of scDb and T cells. Killing of H2122 indeed depended on sotorasib, matched HLA, and KRAS(G12C). Similarly, sotorasib, scDb, and T cells mediated killing of the sotorasib-resistant KRAS(G12C) HLA-A*11-positive cell line H2030. Cryo-EM structural analyses revealed that the HapImmune antibody uses a deep pocket for sotorasib and flat surfaces for HLA, mediating its HLA-dependent recognition of the drug-peptide conjugate and reduced HLA-restriction. Koide and his team have also created HapImmune antibodies to other target–covalent inhibitor pairs to selectively kill tumor cells harboring other mutated disease drivers, proving that this approach is generalizable.
Engineered mRNA and the next generations of bispecific therapeutics- Georg Falck (Mainz, Germany)
Georg Falck introduced LNP-mRNA bispecific drug products consisting of modified mRNA encoding one or more polypeptide chains formulated in lipid nanoparticles (LNPs), which, upon expression in vivo, assemble into a bispecific drug. In contrast to proteins, mRNA therapeutics have the advantage of faster and lower cost production, and a superior pharmacokinetic profile (continuous, extended production in vivo compared with rapid clearance of an injected protein). However, unfavorable features include limited protein expression and heterogeneity of the final drug product due to variable post-translational modifications, such as glycosylation. Design (e.g., caps, 5’ and 3’ UTRs, coding sequences, or polyA), production (e.g., the in vitro transcription process) and purification of the mRNA encoding the therapeutic can all be separately optimized. The most common delivery systems used for mRNA are LNPs, which are composed of structural, ionizable cationic, and stealth lipids, as well as cholesterol. LNPs have a low immunogenicity, and ensure mRNA protection from nucleases, cellular uptake, intracellular release, and effective translation. Since bispecific formats are often prone to aggregation, high protein integrity and monomeric expression need to be confirmed for the LNP-mRNA. Further, in vitro assays are required to verify specific binding, induction of T cell activation and proliferation, and killing of target cells by the expressed bispecific. To prove this point, Falck representatively showed the superior pharmacokinetic profile of the mRNA-encoded CD3/claudin 6-bispecific Fab-(scFv)2, RiboMab02.1, compared to the reference protein in mice. In a non-human primate model, therapeutically relevant serum levels of RiboMab02.1 were detectable for a week after a single dose of BNT142 (LNP-mRNA encoding RiboMab02.1). No adverse events were observed during or after BNT142 treatment. In a therapeutic mouse model, treatment with BNT142 resulted in tumor regression and increased survival. Despite the advances in the field, more needs to be done to narrow down the number of bispecific drug candidates (target, format, and formulation), enable effective delivery via intramuscular or subcutaneous administration, and further enhance mRNA stability.
Antibody–drug conjugates
Next generation of antibody-drug conjugates: New targets, new linkers and new payloads- Takashi Kagari (Tokyo, Japan)
Takashi Kagari opened the session on antibody–drug conjugates (ADC) by providing an overview of the key structural and mechanistic features for this emerging therapeutic class. Features inherent to the drugs themselves include:
(1) the variable domains of the antibody, which define the target selectivity and specificity. Desirable targets are present at > 105 target molecules per cell and undergo internalization. Human or humanized antibodies are preferred to reduce immunogenicity.
(2) the cytotoxic payload molecule, which should be active at sub-nM concentrations, have functional groups that are available to allow conjugation and have biochemical properties conducive to a stable product (such as low hydrophobicity).
(3) the linker and related conjugation chemistry, which are important for payload attachment, including optimization of the drug-to-antibody ratio (DAR), stability, and the release mechanism within the cell.
The eleven currently FDA-approved ADCs utilize a range of options for each of these important features. Kagari categorized them based on cytotoxic payload types, encompassing: DNA alkylating agents (calicheamicin and the PBD dimer tesirine); topoisomerase I inhibitors (DXd and SN-38); and microtubule inhibitors (MMAE, DM1 and DM4) – the most popular payloads. Together these approved drugs target both hematological malignancies as well as solid tumors (10 separate antibody targets), and utilize cleavable and non-cleavable linkers. More than 160 ADCs are in clinical development, with more than 30 different targets; the solid tumor targets HER2,TROP-2 and CLDN18.2 are the most actively investigated. A variety of novel linkers are being examined to optimize the release of drugs to match the characteristics of the targeted tumors, including biochemical conditions (pH), enzymatic access (β-glucuronidase or peptidases), and the presence of degradative co-factors (reduced glutathione). Although cleavable linkers are most widely used, some non-cleavable linkers are being tested. Novel targets are also being investigated, but the field is still dominated by new drugs for existing targets, and although tubulin inhibitors (the predominant payload for approved drugs) continue to be the most common payloads investigated, topoisomerase inhibitors are now the second most common payload type for investigational drugs. Finally, novel directions include bispecific or biparatopic targeting, better tumor-specific cleavage, and combinations of ADC with other drugs or with immunotherapy, portending a broad, deep and bright future for the field according to Kagari.
Combination of antibody-drug conjugates with other agents- Giuseppe Curigliano (Milan, Italy)
Giuseppe Curigliano opened his talk on the activity of agents in combination with antibody–drug conjugates (ADCs) by discussing studies that combined ADCs with antibodies or small molecule inhibitors against the same target. In the Marianne phase 3 study of trastuzumab (T) conjugated to DM1 (T-DM1, trastuzumab emtansine) with or without pertuzumab (P) vs. T + taxane in HER2-positive metastatic breast cancer, the ADC showed no superiority, even when combined with pertuzumab. Two more trials investigated if the ADC can replace T + chemotherapy in the treatment of early breast cancer, but these also failed to prove superiority of the ADC. A multi-arm study of the next-generation ADC T-DXd (trastuzumab deruxtecan) + P is ongoing. Preclinical studies combining T-DM1 with a small molecule HER2 inhibitor (tucatinib) were quite effective, and clinical results appear to slightly extend progression-free survival (PFS) – with a stronger effect in patients with brain metastases – and to increase the objective response rate (ORR). A combination of an ADC with a checkpoint inhibitor is already approved (pembrolizumab plus enfortumab vedotin in urothelial cancer), and the mechanistic rationale of combining these two agents is the synergistic effect of the tumor cell lysis-induced release of antigens with immune stimulation via immune checkpoint inhibition. Surprisingly, T-DM1 + atezolizumab (anti-PD-L1) did not show a significant clinical benefit over T-DM1 alone. T-DXd combinations may be more interesting based on mechanistic studies indicating that T-DXd stimulated immunity and was more active in combination with anti-PD-1. In a clinical study of T-DXd with nivolumab in HER2-positive or HER2-low tumors, the HER2-positive arm outperformed the HER2-low arm in terms of overall response rate and progression-free survival. Interestingly, there was no impact related to PD-L1 status. Another study, the Begonia study, is showing robust and durable responses in the arm combining the anti-Trop2 ADC datopotamab deruxtecan and durvalumab (anti-PD-L1) with no new safety concerns in first-line TNBC. The absence of effects associated with PD-L1 status here led Curigliano to suggest there is no scientific rationale for continuing to require PD-L1 testing in these settings. Combining ADCs with inhibitors of the DNA damage repair pathway is supported by good preclinical evidence, with clinical studies ongoing. Finally, combining HER2-ADCs with endocrine therapy follows the successful path of combining a HER2 antibody therapy with endocrine therapy, as they have non-overlapping mechanisms of action; clinical studies are ongoing.
Tumor agnostic development of antibody-drug conjugates- Barbara Pistilli (Villejuif, Cedex, France)
Cancers originating in different organs may harbor the same molecular alterations and tumor-associated or tumor-specific targets, including some that may have an oncogenic role. This forms the basis of considering tumor-agnostic development of some antibody–drug conjugates (ADCs). Barbara Pistilli started her talk by presenting examples of ADCs that have already demonstrated tumor-agnostic potential. For instance, the ADC trastuzumab emtansine (T-DM1; Kadcyla), which is FDA-approved for the treatment of pretreated HER2-positive breast cancer, has also shown some efficacy against HER2-positive gastric and non-small cell lung cancer (NSCLC). The third-generation ADC trastuzumab deruxtecan (T-DXd, Enhertu) is FDA-approved for the treatment of pre-treated HER2-positive breast and gastric or gastroesophageal junction cancer, and also, since 2022, for the treatment of pre-treated HER2-mutated NSCLC. In a phase 2 trial, T-DXd recently demonstrated an overall response rate of 37.1% and a median progression-free survival of 6.9 months in pre-treated patients with other HER2-positive solid cancers, such as endometrial, cervical, ovarian, bladder, biliary tract, and pancreatic cancer. Analysis of The Cancer Genome Atlas revealed more ADC-targets with expression across multiple different tumor types. Identifying appropriate biomarkers is critical to support tumor-agnostic development. While expression level of the target antigen is associated with response to ADCs such as T-DXd, TROP-2-targeted sacituzumab govitecan, FRα-targeted mirvetuximab soratansine, and others, the optimal minimal target threshold still needs to be determined, and may differ across the tumor types targeted. Different cancer histologies may also affect the activity of the ADCs, as the tumor microenvironment, co-existing molecular alterations, genes and pathways involved in ADC internalization, intracellular trafficking, and linker cleavage may deviate in tumors of diverse organ origin. Finally, the development and use of biomarkers of ADC sensitivity or resistance based on pre- and post-treatment analyses using spatial technology or advanced single-cell transcriptomic technology and correlations with response will add to the needed guidelines and recommendations for the tumor-agnostic use of ADCs.
Optimizing the safety of novel antibody-drug conjugates- Toshio Shimizu (Wakayama, Japan)
With multiple drug approvals over the past few years, ADCs have become an exciting area for further clinical development, especially for solid tumors. However, as reviewed by Toshio Shimizu, safety optimization to improve the therapeutic index is a dominant early hurdle at which multiple developing drugs fail. As such, this is an area of intense investigation for improvement. Toxicity is most commonly associated with on-target, off-site effects, but can also be due to off-target, off-site receptor-mediated internalization of the ADC by FcγR or scavenger receptors, or off-target, off-site non-specific ADC uptake or release of membrane diffusible payloads. Toxicities are also highly related to the payload and are frequently similar to toxicities observed with mechanistically similar chemotherapies. These toxicities are mostly off-target/off-tumor and can be mediated by presence of high levels of macrophages expressing FcγRs or by the local release of the payload and uptake by bystander cells. Approaches to overcoming these issues include modifying the antibody components, conjugation technologies, dosing regimens, and, more recently, inverse targeting. Examples of antibody modifications include (1) a “pro-drug” strategy in which a target-masking agent is released by tumor-localized proteases, (2) bispecific targeting requiring the presence of two different targets or two different epitopes on the same target, leading to more specific binding and internalization, and (3) “silencing” of FcγR binding, which can be achieved by site-specific conjugation. Finding the right type of linker can be a key to finding the proper balance (the yin and yang) of stability and release, and hence, efficacy versus toxicity. The linker type can also affect the drug-to-antibody ratio (DAR), which may not always follow a “more is better” rule, due to enhanced liver clearance, more off-target hydrophobic effects, or interference with target binding. More extensive dose optimization studies (e.g., additional patients randomized to receive doses near the dose identified by conventional 3 + 3 designs) can also provide a clearer understanding of the dose–toxicity relationship. Other dose optimization opportunities that impact toxicity include dose fractionation, treatment-guided adjustments, and capping maximal exposure. Finally, Shimizu described a technology just entering clinical trials known as Inverse Targeting, in which a small antibody fragment that binds to and neutralizes the activity of the payload is co-injected to provide blocking of payload activity – hence blocking of systemic toxic effects in the blood - where the concentration of the ADC is high, but allows activity in the tumor, where the concentration of the blocking small antibody fragment is low. Beyond modifying the drug, genomic analysis (identification of genetic variants with high sensitivity to particular toxicities) and analysis of tumor biomarkers affecting activity or toxicity brings in patient selection as a tool to optimize the therapeutic index.
Targeted interleukins
Phase I dose-escalation trial with tumor-targeted interleukin-12 (IL12-L19L19) in patients with solid tumors- Giuseppe Curigliano (Milan, Italy)
Giuseppe Curligliano presented the preliminary results of the dose escalation part of a phase I clinical trial testing a tumor-targeted form of IL-12 created by fusing an oncofetal fibronectin (EDB)-targeting antibody (L19) to IL-12. Pan-tumoral, moderate/high expression of EDB would potentially deliver IL-12 to the TME to activate/reactivate immunogenicity. The IL-12/L19 immunocytokine was introduced intravenously in a standard 3 + 3 design at doses ranging from 0.5 to 16 ug/kg. IL-12/L19 was administered once a week for 8 weeks, followed in some patients by biweekly dosing to a maximum of 48 weeks. Patients were required to have solid tumors for which checkpoint therapy has been approved, and to have been previously successfully treated with immunotherapy (at least 3 months free from disease progression following treatment). Among 22 evaluable patients, 3 dose-limiting toxicities (DLTs) were observed, and a tolerable dose limit of 8 ug/kg was established. IL-12/L19 had a short half-life in circulation, and studies with L19 alone demonstrated prolonged retention in the tumor, as designed. Multiple serum biomarkers (IFNγ, IP-10, neopterin, IL-6) were followed, all of which showed increases 24 hours after injection, suggesting reactivation of the immune response. In an exemplary case, a patient with TNBC received IL-12/L19 treatment (the 8 weekly doses only) and showed a partial response that lasted for 4 months. Encouragingly, 10 of 22 patients demonstrated stable disease following treatment.
Beyond PD-1/PDL-1: Engineered PD-1-cis IL2 to overcome T cell exhaustion- Ignacio Melero (Pamplona, Spain)
IL-2 is a potent cytokine with a key stimulatory role for NK cells, antigen-activated CD4+ and CD8+ T cells, and regulatory T cells. The multi-subunit IL-2 receptor (αβγ) is expressed on both immune, and some important toxicity-inducing non-immune cells, in different compositions (αβγ or βγ), conferring different affinities for IL-2. Thus, as summarized and presented by Ignacio Melero, taming this supercharging cytokine has been of intense interest and significant complexity. One key approach has been to target variants of the cytokine to the low-affinity (0.5 nM) βγ receptor rather than the high-affinity (12 pM) αβγ receptor, to favor activation of effector T cells, rather than endothelial cells and immunosuppressive regulatory T cells, both of which express the high-affinity receptor. Such variants, known as IL-2v, can be targeted to certain cells/tissues by linkage to a targeting domain, such as an scFv. Targeting IL-2v to fibroblast activation protein (FAP), expressed significantly in the tumor microenvironment, showed increased tolerability and specific expansion of CD8+ T cells and NK cells, but not Tregs. However, only moderate objective responses were observed, likely due to exhaustion of T cells within the TME. The chronic LCMV (Clone 13) model of T cell exhaustion proved valuable in an attempt to overcome such a limitation. Melero showed that using the βγ receptor-targeted IL-2v led to weak antigen-specific CD8+ T cell activation, but that by targeting IL-2v in cis to PD-L1 with a unique dimeric PD-1 moiety bearing a silenced Fc-domain, enhanced T cell activation, an increased CD8/CD4 ratio, especially in tumors, and therapeutic efficacy were observed. Improved effectors were induced from a unique population of stem-like CD8+ T cells (confirmed by single-cell RNA sequencing), bypassing induction of conventional exhausted CD8+ T cells. Improved tumor control was observed in different subcutaneous, orthotopic, or autochthonous murine tumor models. This cis-targeted IL-2v is currently entering Phase I clinical studies.
Adoptive cell therapy
Armored effector cells for treatment of solid tumors- Michael O’Dwyer (Galway, Ireland)
Despite success in hematologic malignancies, Michael O’Dwyer reviewed the multiple obstacles faced by CAR T cells in achieving success against solid tumors. A large and heterogeneous tumor mass harbors conditions that prevent good T cell infiltration, and factors that suppress and exhaust infiltrating T cells. Among the T cell inhibitory ligands expressed on tumor cells, Fas ligand (Fas-L) limits activity and persistence by inducing T cell apoptosis. In some cells, Fas-L engagement alone is not sufficient to induce apoptosis, but requires cooperation with an intrinsic pathway in mitochondria – a pathway that can be blocked by Bcl-xL or Bcl-2. Work from Phil Greenberg’s lab previously showed that a Fas ectodomain:4-1BB signaling domain chimera expressed in adoptively transferred T cells could functionally deliver a stimulatory signal in the presence of tumor-expressed Fas-L, maintaining T cell viability and antitumor activity in mouse models. Interestingly, patients with multiple myeloma who were treated with BCMA-CAR T cells showed a positive correlation between higher Bcl-xL production and clinical response, and it was known that canonical CD28 co-stimulation of T cells led to induction Bcl-xL expression and maintenance of viability under conditions of Fas exposure and IL-2 deprivation. These observations led to the idea of overexpressing Bcl-xL in CAR T cells as a way to prevent the negative effects of tumor-expressed Fas-L. “Armored” Bcl-xL-expressing CAR T cells demonstrated better viability and maintenance of cytotoxicity in response to chronic antigen exposure in vitro, a phenotype characterized by less exhaustion, higher TCF-1 expression, a higher proportion of central memory and precursor exhausted cells, and a stronger in vivo antitumor effect. NK cells also have a critical role in both direct antitumor activity and in activation of adaptive immunity, but, similar to T cells, become impaired in the tumor microenvironment. Recent work has shown that IL-15 stimulation is a partially effective approach to overcoming such functional impairment in NK cells, and O’Dwyer described how knockout of CISH, an intracellular checkpoint in the IL-15 stimulation pathway, could enhance the impact of IL-15 stimulation in both T cells and NK cells. IL-15 knockin and CISH knockout in human NK cells led to tumor cell killing in a human tumor spheroid model. TGFβ is also a highly immunosuppressive factor in the TME, and CISH knockout synergizes with TGFβR2 knockout. At the end of his talk, O’Dwyer described how the combination of knockout of TGFβR2 and CISH together with knockin of IL-15 produced highly active CAR NK cells with high cytotoxic efficacy in the spheroid assay, even in the presence of TGFβ.
TIL therapy in solid tumors- Ronnie Shapira-Frommer (Ramat Gan, Israel)
It took 36 years since the first seminal paper describing the clinical activity of tumor-infiltrating lymphocyte (TIL) therapy was published, but this year marked the first FDA approval of TIL therapy. Ronnie Shapira-Frommer summarized the experience of TIL therapy at her institution over the past approximately 17 years. In contrast to selecting subpopulations of TILs, which can extend the production time, beginning in 2008, Shapria-Frommer’s TIL product derived from bulk expansion of all TILs, which resulted in younger TILs with longer telomeres, a more proliferative phenotype, and tumor reactivity similar to selected TILs. Of the approximately 100 patients over 8 years treated with this unselected TIL product, the response rate averaged about 30%, with all complete responders and many partial responders still alive. However, many issues persist with TIL therapy, including toxicity of lymphodepleting and high-dose IL-2 therapy, the dropout rate of 30% (mostly due to disease progression prior to product availability), and high cost, all of which limit TIL product use, particularly compared to off-the-shelf checkpoint blockade therapies. Moreover, the checkpoint inhibitor therapy era led to patients initiating TIL therapy only after being treated with one or more lines of checkpoint blockade, often over multiple years until ultimately becoming refractory. Both Shapira-Frommer’s group and others observed a concomitant dramatic drop in effectiveness of TIL therapy. Turning to how to make TIL therapy more successful for the 70% of melanoma patients who will eventually transition from checkpoint blockade to TIL therapy, one clue was an observed increase in markers of exhaustion and a reduction in markers for stemness of the TILs in resistant patients. A second clue came from examining the treatment history of patients in the large published TIL therapy trials, which suggested that selection of patients before the development of terminal resistance was likely important. Indeed, instituting more stringent patient selection over the past two years has led to an increase in TIL effectiveness. Other ongoing approaches being investigated in the clinic to improve outcomes include selection of CD137+ T cells (indicative of neoantigen-specific T cells), fecal microbiota transplant prior to tumor harvesting for TIL production and prior to TIL injection, and knockout of CISH, an intracellular checkpoint target. Shapira-Frommer’s lab is also investigating knockout of other targets that can confer intrinsic resistance. Finally, while successful in establishing TILs from ovarian cancer patients and initiating a clinical trial, in the two years that the trial has been open, no patients have been enrolled due to the complexity of considering prior and post TIL-therapy chemotherapy treatment options, demanding protocol amendments.
EGFR-CAR NK against rhabdomyosarcoma and glioblastoma- Evelyn Ullrich (Frankfurt am Main, Germany)
CAR T cell therapies have revolutionized the adoptive cell therapy field, but many challenges remain, particularly for solid tumors; these challenges include toxicity, individualization, and antigen loss. As summarized by Evelyn Ullrich, CAR NK cells offer an alternative, capitalizing on a high intrinsic killing capacity, the ability to recognize “missing self” (recognizing tumors with HLA downregulation) and the opportunity for an off-the-shelf product. Indeed, cord blood-derived CD19 CAR NK cells expressing IL-15 have shown impressive clinical results against B cell malignancies, including better persistence in clinical responders versus non-responders. The NK-92 NK cell line provides a convenient source compared to naturally occurring NK cells, but also exhibits some potentially advantageous phenotypic differences, including absence of CD16 (no ADCC potential) and a fixed repertoire of inhibitory and stimulatory receptors. Preclinical studies in glioblastoma supported an initial clinical trial of HER2-CAR NK-92 cells delivered intracranially via a port immediately following tumor resection, which demonstrated safety and promising activity. The intracranial port allows repeated infusions, which may be important, as NK-92 cell products require irradiation prior to injection and so have limited persistence. To overcome immunosuppression, checkpoint blockade with an anti-PD-1 antibody was added to the treatment regimen, with an encouraging progression-free survival outcome. The trial design for the checkpoint inhibitor-treated cohort was to biopsy a relapsed tumor, inject the HER2-CAR NK-92 cells and anti-PD-1 antibodies into the relapsed tumor, and then surgically resect the tumor and treat with HER2-CAR NK cells again, allowing for immunohistochemistry (IHC) analysis of the tumor prior to and after CAR treatment. IHC revealed an increase in CD4+ and CD8+ T cells following treatment, which inversely correlated with HIF1α levels, suggesting knockout of HIF1α might be a useful further opportunity. Turning back to primary NK cells to produce CAR NK cells, Ullrich described the preparation of primary CAR NK cells targeting EGFR/EGFRvIII (using the scFv derived from cetuximab). These cells were cytotoxic to multiple EGFR-expressing cell lines in vitro, slowed EGFR+ GBM tumor growth in a subcutaneous xenograft model, and were, in combination with local radiotherapy (to promote immune cell migration into the tumor) and IL-2, active against EGFR-expressing rhabdomyosarcoma in a xenograft model, expanding the targetable disease range.
Resistance to CART therapy- Patrizia Porazzi (Philadelphia, United States of America)
Despite the clinical and commercial success of approved CAR T cell products targeting CD19 and BCMA, significantly fewer than half of patients durably respond, and these therapies only prevent or delay a small proportion of potential cancer deaths. Patrizia Porazzi reviewed how CAR T cell therapy could be effectively expanded in breadth and effectiveness. A clear issue for solid tumors is identifying antigens with the proper features, including sufficient overexpression, lack of expression on other vital tissues/organs, and, ideally, having some role in oncogenesis or tumor survival to prevent antigen loss. Porazzi briefly summarized logistic barriers to effective CAR therapy, such as low lymphocyte counts due to prior therapies and various manufacturing issues (manufacturing failure, long manufacturing times that result in some patients progressing during product preparation, and high cost of manufacturing), but mostly focused on approaches to improve CAR T cell quality and response to the suppressive tumor immune microenvironment. CAR T cell dysfunction is commonly caused by chronic antigen exposure, leading to an exhausted phenotype with poor cytotoxic capacity compared to the more desirable highly cytotoxic central memory phenotype. Knockout or knockdown of intracellular inflammatory mediators or local slow-release delivery of gel-encapsulated CAR T cells can prevent chronic antigen exposure-induced loss of function. Alternatively, Porazzi described removing negative regulators of T cell function. For example, removal of surface-expressed CD5 in mesothelin CAR T cells enhanced in vivo tumor cell killing, overall survival, and T cell persistence in an animal model of pancreatic cancer. The tumor microenvironment presents multiple barriers to effective CAR T cell therapy. A physical barrier arises from the desmoplastic stroma, induced primarily by cancer-associated fibroblasts. Various extracellular matrix-degrading strategies are under development, and one example treatment with a hyaluronidase-secreting oncolytic virus in combination with a CAR T cell product is currently being clinically tested. Biologically, TGFβ is a highly immunosuppressive cytokine, and trials with knockdown or with a dominant negative TGFβ receptor gene to reduce the impact of intratumoral TGFβ are underway. Finally, Porazzi described a multi-dimensional approach to impacting CAR T cells, tumor cells, and the tumor microenvironment all at the same time, by epigenetic modulation with tazemetostat (FDA-approved for the treatment of follicular lymphoma and epithelioid sarcoma), an inhibitor of the histone methylase EZH2. CD19-CAR T cell therapy together with EZH2 inhibition led to enhanced tumor control in vivo. Inhibition of EZH2 enhances CAR T cell expansion, primarily of non-Treg CD4+ T cells with a naive or early memory phenotype (CAR T cell effect). RNA sequencing of tumor cells from EZH2-inhibitor treated mice showed upregulation of genes involved in B cell stimulation, interferon response, and apoptosis (tumor cell effect), and a novel assay assessing cell:cell interactions showed enhanced adhesion of CAR T cells and inhibitor-treated tumor cells (TME effect). In vitro, tazemetostat enhanced T cell killing of Ewing sarcoma cells and HER2-CAR T cell killing of HER2-expressing prostatic adenocarcinoma or ovarian cancer cells.
By Ute Burkhardt, Ed Fritsch, and Lauren Hitchings



