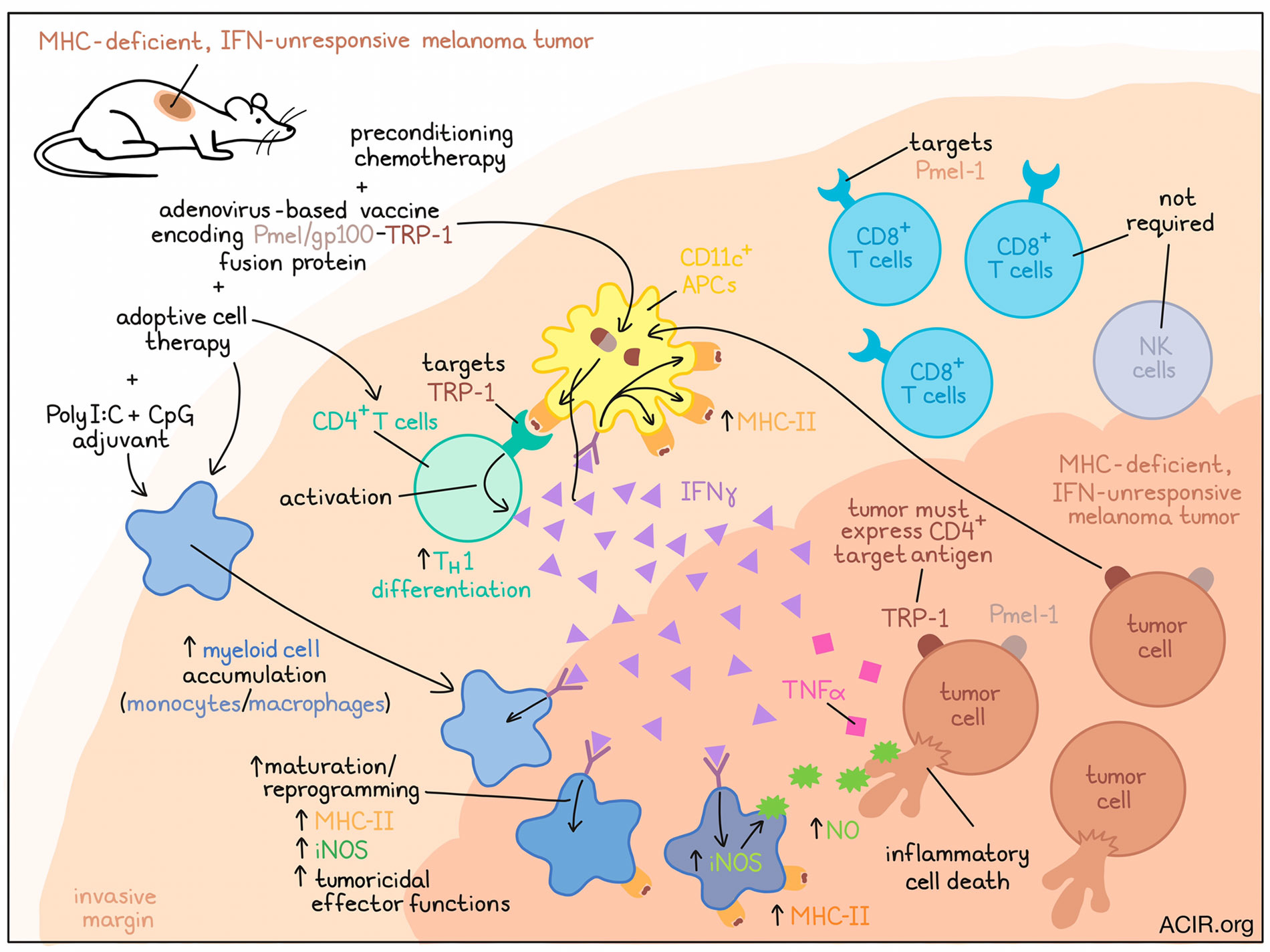
Investigating the contributions of CD4+ T cells to antitumor immunity, Kruse, Buzzai, Shridhar, and Braun et al. unraveled a mechanism through which a small number of CD4+ T cells can eradicate MHC-deficient, IFNγ-unresponsive tumors that escape direct CD8+ T cell targeting. Their results, recently published in Nature, show that, in concert with innate immune stimulation, CD4+ T cells cluster at tumor invasive margins, where they are activated by MHC-II+CD11c+ antigen-presenting cells, reprogram the tumor-associated myeloid cell network, and support inflammatory cell death through IFNγ, TNFα, and iNOS.
To begin, the researchers evaluated samples and data from skin metastases of melanoma, and found that downregulation of MHC-I was relatively common, and was associated with the absence of CD8+ T cells and poor response to immune checkpoint blockade. Meanwhile, MHC-II was almost always restricted to stromal and antigen-presenting immune cells at invasive margins, and was also associated with infiltrating CD8+ T cells.
To directly study the behavior of CD4+ and CD8+ T cells in tumors, the researchers fluorescently tagged TRP-1-targeting CD4+ T cells and Pmel-1-targeting CD8+ T cells. These cells were then used for adoptive cell transfer in B16 melanoma-bearing mice (which endogenously express both target epitopes) in combination with preconditioning chemotherapy, an adenovirus-based vaccine encoding a Pmel/gp100-TRP-1 fusion protein, and adjuvant injections with polyI:C and CpG. Compared to CD8+ T cells, CD4+ T cells expanded much less efficiently, but were equally capable of eradicating established tumors. Preconditioning chemotherapy and innate immune stimulators were essential to antitumor efficacy, contributing to the differentiation of transferred and endogenous CD4+ T cells towards a TH1 phenotype, and to the prevention of Treg accumulation. Both ACT and innate immune stimulation each independently increased the myeloid immune infiltrate (consisting predominantly of monocytes and macrophages), and their effect on monocyte accumulation was stronger in combination.
As most human melanoma cells do not express MHC-II, the researchers next investigated whether TRP-1-targeting CD4+ T cells could control Ciita-knockout (MHC-II-deficient) HCmel12 tumors. In vitro, TRP-1-targeting CD4+ T cells could directly respond to MHC-II-expressing HCmel12 cells in an antigen-specific manner, but were more efficiently activated indirectly by MHC-II+ dendritic cells pulsed with HCmel12 cell lysates. In vivo, TRP-1-targeting CD4+ T cells could eradicate MHC-II-deficient tumors, as long as TRP-1 was expressed. While some bystander killing was observed in MHC-II-deficient tumors with heterogeneous TRP-1 expression, the outgrowth of Trp1-KO cells could not be fully controlled.
Given that HCmel12 cells only express MHC-I and MHC-II upon exposure to IFNs, the researchers generated Jak1-KO HCmel12 cells to disrupt IFN signaling and MHC expression and study the effects of CD4+ T cells in isolation. When NK cells were depleted, and with no CD8+ T cell activity, adoptively transferred TRP-1-targeting CD4+ T cells alone eradicated established tumors. In this IFN-unresponsive, MHC-deficient model, both TRP-1-targeting CD4+ T cells and Pmel-1 CD8+ T cells substantially increased the number of myeloid cells in tumors, though the increased immune cell infiltrate was less pronounced in Jak1-KO versus control tumors. Similar results were observed in a model in which OVA was the target antigen for OT-I and OT-II cells.
Next, Kruse, Buzzai, Shridhar, and Braun et al. then proposed that CD4+ effector T cells might be activated in tumor tissues by MHC-II+ antigen-presenting cells, whereas CD8+ T cells need to be activated by MHC-I-restricted antigens on tumor cells. This effect was demonstrated using tumor imaging of amelanotic Tyr-KO tumors. In control Tyr-KO tumors, low numbers of CD4+ T cells were identified in invasive margins, while high numbers of CD8+ T cells infiltrated both invasive margins and the tumor center. In MHC-deficient tumors, a low number of CD4+ T cells and a high number of CD8+ T cells were again identified at the invasive margin, but CD8+ T cells remained highly motile and did not accumulate in the tumor center. Similar results were observed using an OVA-based tumor immunity model.
Using fluorescently tagged Tyr-KO HCmel12 tumor cells in mice with tagged antigen-presenting cells, the researchers were able to visualize tagged CD4+ T cells stopping and preferentially engaging with tagged MHC-II+ antigen-presenting cells within tumor invasive margins. The antigen-presenting immune cells located near CD4+ T cells upregulated the expression of MHC-II exclusively, consistent with the notion that CD4+ T cells had been locally activated and were secreting IFNγ. These effects were not observed in Trp1-KO tumors, or when MHC-II blockade was used, suggesting that they occur in an antigen recognition-dependent manner.
To better understand how so few CD4+ T cells could so strongly control tumor growth, the researchers evaluated CD11b+Ly6G- tumor-infiltrating mononuclear phagocytes from untreated mice and mice treated with the CD4 ACT protocol. This revealed strong activation of IFN-response genes following treatment, and an influx of immature monocytes that shifted towards more IFN-activated monocyte/macrophage cell states, including MHC-II antigen-presenting, iNOS-expressing, and tumoricidal states. Further, CD4+ T cells showed synergy with the innate immune stimulators - an effect found to be due to CD4+ T cell production of IFNγ, which contributed to enhanced iNOS expression and tumoricidal monocyte effector qualities.
Investigating exactly which factors in the TME contribute to tumor cell death, the researchers found that iNOS was essential to antitumor efficacy in Jak1-KO (IFN-unresponsive, MHC-deficient) HCmel12 tumors, but not in Ciita-KO (IFN responsive, MHC-II-deficient) tumors. CCR2+ monocytes were also more critical than neutrophils to this antitumor effect, suggesting an important role for iNOS-expressing monocytes and macrophages. Investigating a recently described mode of inflammatory cell death driven by the concerted action of IFNγ, TNF, and nitric oxide, the researchers found that the nitric oxide donor SNAP effectively induced death in both Ciita-KO (IFN-responsive) and Jak1-KO (IFN-unresponsive) HCmel12 cells in vitro. Meanwhile, the combination of IFNγ and TNF was only sufficient against Ciita-KO (IFN-responsive) HCmel12 cells. Similar results were observed using human cells, supporting the hypothesis that in IFN-responsive melanoma, IFNγ sensitizes cells towards TNF-induced cell death, while in IFN-unresponsive melanoma, myeloid cell-derived nitric oxide contributes to efficient inflammatory cell death.
Overall, these findings suggest that, supported by pre-conditioning and innate immune stimulation, a relatively small number of CD4+ T cells can eradicate MHC-deficient, IFNγ-unresponsive tumors that otherwise escape CD8+ T cell detection. This effect is based on CD4+ T cell activation by antigen-presenting cells in tumor invasive margins, followed by production of IFNγ, which contributes to monocyte/macrophage reprogramming and inflammatory cell death.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, co-first authors Bastian Kruse and Anthony C. Buzzai answered our questions.

What was the most surprising finding of this study for you?
The first surprising result came to us when we observed that very few CD4+ T cells could control IFN-unresponsive tumours, which resisted CD8+ T cell immunity. At the time, we did not understand how CD4+ T cells could control these tumours so effectively, which led us to discover that the spatial distribution of the CD4+ T cells fundamentally differs when compared to CD8+ T cells. This brought us to our second astounding finding that the CD4+ T cells do not infiltrate into tumour tissues, but rather, eradicate melanomas remotely from the outside-in by interacting with recruited myeloid cells.
What is the outlook?
We want to further explore how CD4+ T cells and innate immune stimulators synergise to control tumours, since it is not entirely understood how their interplay sensitises cancer cells to death. We hope our findings bring CD4+ T cells into the spotlight to ignite further research into their contribution towards antitumor immunity. We believe that exploiting CD4+ T cell effector functions will benefit cancer patients in the future.
What was the coolest thing you’ve learned (about) recently outside of work?
BK: I recently learned that individual orcas have signature calls to identify each other. Marine biologists believe that these unique calls are their names.
ACB: You should only play 20% of your starting hands in poker… I learnt this the hard way.



