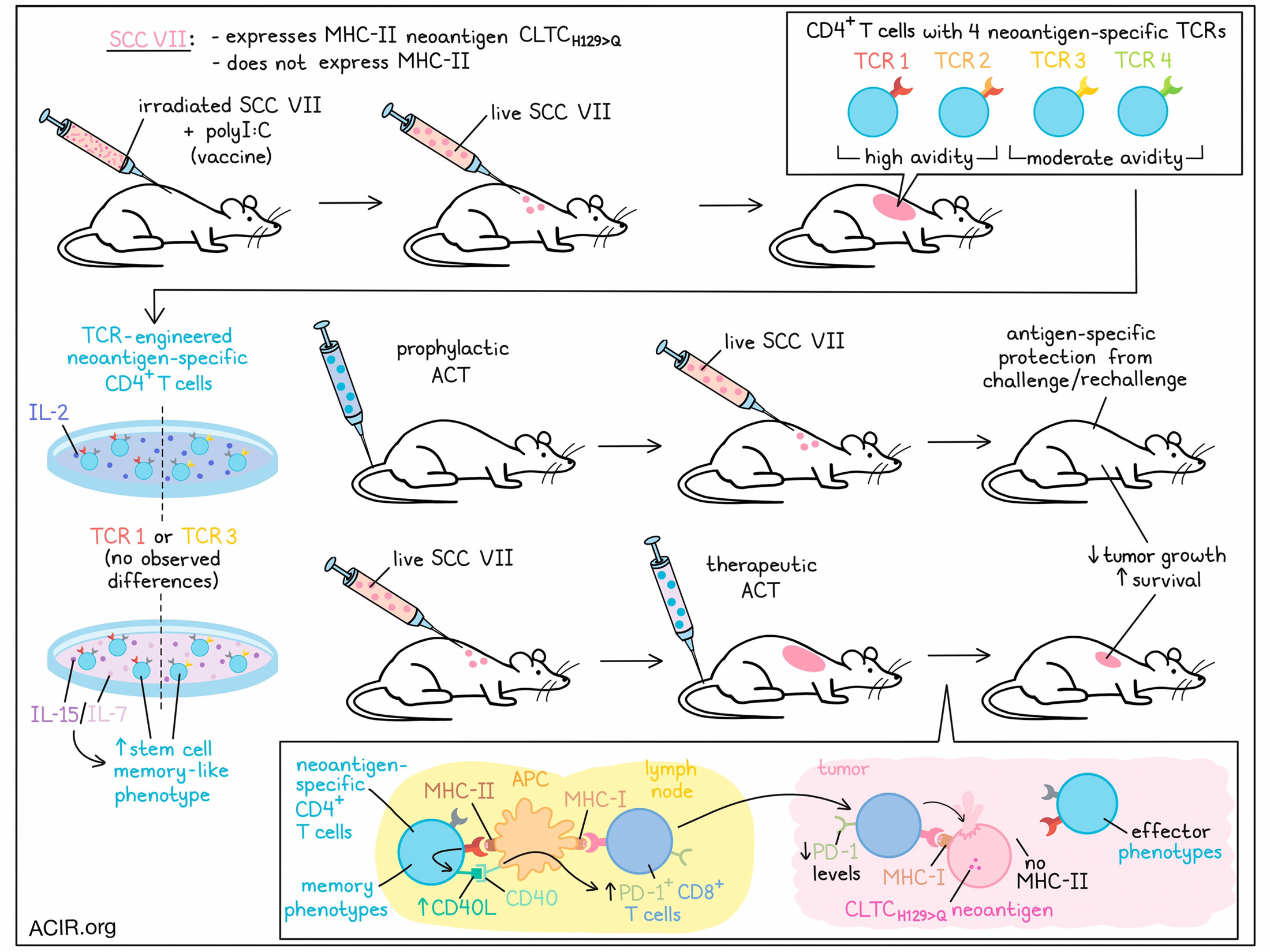
CD4+ T cells have been shown time and time again to play roles in antitumor immunity, but the exact mechanism and factors that influence their effects remain unclear. Previous research in mice identified a specific CD4+ T cell response to CLTCH129>Q – a validated neoantigen target expressed by the MHC-II-deficient squamous cell carcinoma tumor model (SCC VII). Furthering this research, Brightman et al. recently identified four distinct TCR clonotypes recognizing this target with different avidities, and evaluated and optimized the capacity of CLTCH129>Q-specific CD4+ T cells to contribute to antitumor immunity. Their results were published in Nature Immunology.
To begin, Brightman et al. used mice that were vaccinated with irradiated SCC VII cells and adjuvant polyI:C and were protected from SCC VII live tumor challenge to isolate an expanded pool of CLTCH129>Q-specific CD4+ T cells. From there, the researchers identified and validated four distinct TCRs consisting of 3 TCR β clonotypes paired with four α-chains. TCR 1 and TCR 2 shared the same TCR β-chain and nearly identical α-chains that differed by only a single amino acid. Further, TCRs 1 and 2 showed notably (~10x) higher avidities than TCRs 3 and 4, though unlike results previously seen in CD8+ T cells, this was not found to correlate with differences in proximal TCR signaling.
To study the expansion and activation of CLTCH129>Q-specific CD4+ T cells in vivo, the researchers transferred equal numbers of retrovirally transfected, labeled CD4+ T cells expressing either TCR 1 or TCR 3 into naive mice. When the mice were subsequently challenged with SCC VII, both subsets responded to antigen recognition, expanding equally, expressing similar levels of the acute activation marker CD69, and upregulating PD-1 to a similar extent.
While SCC VII does not express MHC-II, even under inflammatory conditions, adoptive transfer of activated CD4+ T cells expressing TCR 1 protected mice from tumor challenge 1 day later. This protection was antigen-specific, as the same protection was not observed when mice were treated with activated polyclonal non-specific CD4+ T cells. Mice that were protected from the initial challenge were also protected from rechallenge at 30 days, suggesting that CLTCH129>Q-specific CD4+ T cells mediates durable immune memory. These results were dependent on the number of cells transferred, as mice that received lower cell doses showed a reduced survival benefit compared to those receiving higher cell doses.
Given that CLTCH129>Q-specific CD4+ T cells cannot directly recognize neoantigens on tumor cells due to the lack of MHC-II on SCC VII tumor cells, the researchers hypothesized that the observed antitumor effects were likely mediated through CD8+ T cell help. Indeed, CD8+ T cell depletion prior to the adoptive transfer of neoantigen-specific CD4+ T cells abrogated their protective effect. Based on recent evidence that CD4+ T cells provide help through CD40L-dependent licensing of cDC1s, the researchers evaluated CD4+ T cells expressing TCR 1 for CD40L expression and found that it was upregulated upon in vitro exposure to peptide-pulsed splenocytes. In vivo, CD40L-blocking antibodies administered on the day of tumor implantation and again 2 days later abrogated antitumor immunity induced by neoantigen-specific CD4+ T cells. Again, there was no difference between TCR 1 and TCR 3.
Next, Brightman et al. investigated the potential efficacy of CLTCH129>Q-specific CD4+ T cells against large, established tumors. In this setting, neoantigen-specific CD4+ T cells proliferated in tdLNs and infiltrated tumors, but failed to improve survival. In an effort to enhance their potency, the researchers tried culturing the cells in IL-15 and IL-7 prior to transfer, rather than the standard IL-2, as this strategy has been shown to induce a less differentiated stem cell memory-like state and enhance ACT in studies with CD8+ T cells. CLTCH129>Q-specific CD4+ T cells cultured in IL-15/IL-7 showed much greater expansion in peripheral blood and accumulation in tumors and tdLNs compared to cells cultured in IL-2, and effectively delayed tumor growth and enhanced survival. Results were similar between CD4+ T cells expressing TCR 1 or TCR 3.
Taking a closer look at CLTCH129>Q-specific CD4+ T cells in different locations, the researchers found through gene expression analysis that cells in the tdLNs differentially expressed markers of memory T cells, while those in tumors differentially expressed genes associated with effector functions, including TH1 cytokines, TCR signaling pathway components, and cytotoxicity genes. In line with this, flow cytometry revealed that while tumor populations contained mostly effector memory T cells, tdLNs contained various memory subpopulations, including effector memory, central memory, and stem cell memory-like populations. Notably, a unique memory population was observed when cells were cultured in IL-15/IL-7 that was not observed when cells were treated with IL-2. These results suggest that the lymph nodes act as a reservoir for transferred TSCM cells, which give rise to more differentiated subsets that traffick to tumors.
To evaluate the impact of therapeutically transferred CLTCH129>Q-specific CD4+ T cells (cultured in IL-15/IL-7) on CD8+ T cell immunity, the researchers measured PD-1 expression. While there was no change in the percentage of host CD8+ T cells expressing PD-1 in the tumor, the levels of PD-1 were lower, suggesting reduced terminal exhaustion. In tdLNs, the researchers found a significant increase in PD-1+CD8+ T cells, which correlated with the number of transferred cells, suggesting increased priming. When CLTCH129>Q-specific CD4+ T cells were transferred into tumor-bearing mice that were recently depleted of CD8+ T cells, some initial antitumor effects were observed, but longer-term tumor growth delay was lost. Together, these results suggests that neoantigen-specific TSCM-like CD4+ T cells are actively involved in the priming of tumor-specific CD8+ T cells in tdLNs.
Overall, these findings suggest that CLTCH129>Q-specific CD4+ T cells induce antitumor immunity against SCC VII through CD40L, resulting in enhanced priming of CD8+ T cells. Antitumor effects were apparent regardless of TCR avidity, and could be enhanced by culturing engineered neoantigen-specific T cells in a cocktail of IL-15/IL-7 to induce a stem cell memory-like phenotype that enhances in vivo T cell expansion and induces durable tumor control.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Spencer Brightman and lead author Stephen Schoenberger answered our questions.
What was the most surprising finding of this study for you?
SB: We were surprised to see that CD4+ T cells expressing the T cell receptors with moderate avidity were similarly functional to those expressing higher-avidity receptors in vivo in terms of expansion, antitumor efficacy, and surface marker phenotype. We were also surprised to find that the therapeutically transferred T cells maintained their stem-like phenotype in the tumor-draining lymph node several days after transfer.
SS: I've been working on the mechanism of CD4+ T help for CD8+ T cells for a number of years and so was gratified, rather than surprised, that it also regulates neoantigen-specific responses. I was, however, surprised that there appears to be little difference in the efficacy of high- versus moderate-affinity CD4+ T cells, and that their therapeutic capacity involves CD8-dependent and -independent activities.
What is the outlook?
SB: We hope that these findings will influence the field by increasing the appetite for immunotherapies focusing on CD4+ T cells in addition to cytotoxic CD8+ T cells, which have been the main focus of immunotherapies to date. Further studies to advance these findings ought to determine the broad cellular and transcriptomic alterations to the tumor microenvironment induced by adoptive transfer with stem-like neoantigen-specific CD4+ T cells.
SS: Ah, this is where it gets interesting! Current adoptive cell therapy involves engineered T cells specific for a single target antigen expressed by the tumor, and it's been observed in patients that this kind of approach can be rendered ineffective if the tumor loses or modifies its presentation of that one antigen. Our work shows that effective adoptive immunotherapy can be achieved by targeting antigen-presenting cells that cross-present tumor antigen, and this enhances activation, not killing. These activated APC can then launch an army of cytotoxic CD8+ T cells specific for many tumor antigens rather than only one. We are ready to translate this to the clinic now, and if successful, this approach could inform a new approach to immunotherapy.
What was the coolest thing you’ve learned (about) recently outside of work?
SS: I've recently become an "empty nester" and have been enjoying all the opportunities that change has compelled: more travel, less shopping, and a quieter life.



