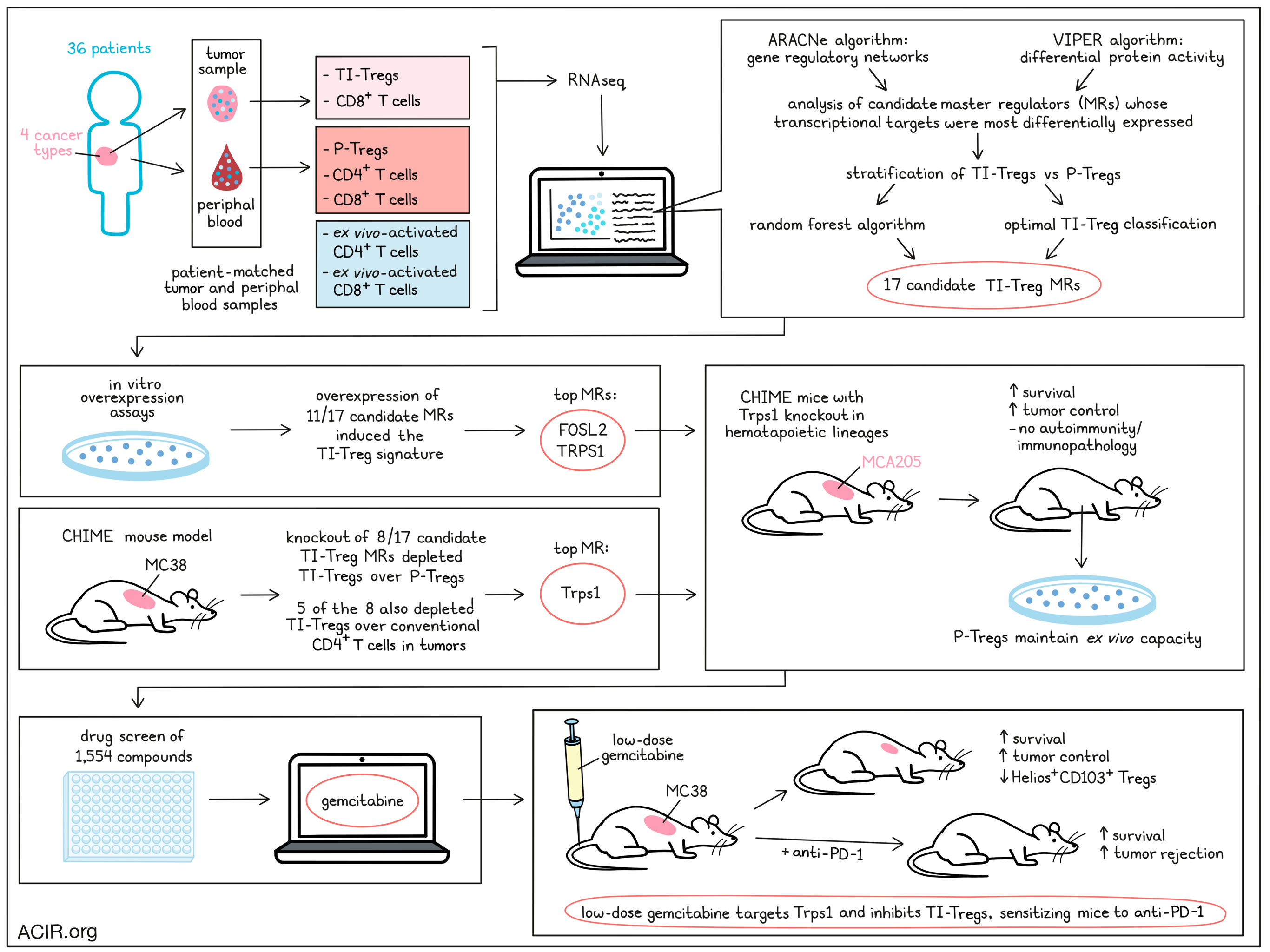
Tregs are an attractive target in cancer immunotherapy, as they play a strong role in inhibiting antitumor immune responses. However, targeting Tregs can also disrupt the system of checks and balances that prevents harmful autoimmune responses. In search of strategies for targeting Tregs that infiltrate tumors (TI-Tregs) without disrupting Tregs in the periphery (P-Tregs), Obradovic, Ager, and Turunen et al. developed a method of identifying master regulatory (MR) proteins that drive infiltration and retention of TI-Tregs. Screening drugs that specifically targeted these TI-Treg MRs revealed a previously unknown effect of gemcitabine in depleting Tregs specifically in the tumor microenvironment (TME). Their results were presented in Cancer Cell.
The researchers began by collecting matched tumor and blood samples from 36 patients with glioblastoma, bladder adenocarcinoma, clear cell renal carcinoma, and prostate adenocarcinoma – cancers that are less represented in existing datasets. Using flow cytometry, they sorted TI-Tregs and P-Tregs, along with peripheral blood CD4+ and CD8+ T cells, and tumor-infiltrating CD8+ T cells. Naive peripheral CD4+ and CD8+ T cell were also collected and activated ex vivo as additional controls for activation. RNAseq profiles were then generated using Illumina sequencing, and the ARACNe and VIPER algorithms were applied to generate a TI-Treg-specific gene-regulatory network and evaluate differential protein activity, respectively. Activity-based cluster analysis identified a distinct cluster of tumor-infiltrating T cells, and interestingly, TI-Tregs were found to have a relatively tumor type-agnostic transcriptional state. While gene expression data alone could not distinguish between TI-Tregs and P-Tregs, analysis of candidate MRs whose transcriptional targets were most differentially expressed nearly perfectly stratified TI-Tregs and P-Tregs across all tumor types.
Next, the team used the random forest algorithm to identify the 15 most discriminative candidate MRs among those differentially expressed in TI-Tregs. Seven candidate MRs were also identified for optimally classifying TI-Tregs vs. other control subpopulations, 2 of which were not included in the previously identified candidate MRs.
To validate the 17 total candidate TI-Treg MRs, many of which had known roles in Treg function or suppression of T cell activation, the researchers used in vitro overexpression assays to determine whether any of the individual MRs could induce naive human P-Tregs to adopt a TI-Treg state. Following scRNAseq and unsupervised clustering, the researchers identified a cluster of cells enriched for the TI-Treg signature, which consisted of all 17 candidate TI-Treg genes. Analysis revealed that 11 out of the 17 candidate MRs were sufficient to induce changes in the MR module and shift the overall transcriptional signature to that of TI-Tregs, with all MRs concurrently active at the single-cell level. The strongest enrichments were seen with the MRs FOSL2 and TRPS1.
Forin vivo validation, Obradovic, Ager, and Turunen et al. used the CHIME mouse model to perform pooled CRISPR-Cas9 screens and assess whether an immune cell lineage-specific knockout of MRs could deplete TI-Tregs without impacting P-Tregs. In Cas9-tolerized mice with HSC-reconstituted immune systems, most tranduced Tregs harbored a single sgRNA perturbation. Upon implantation of MC38 tumors, positive control genes were depleted in Tregs as expected, and 8 MRs presented with significantly depleted sgRNAs in TI-Tregs vs. P-Tregs. Among these, 5 were also significantly depleted in TI-Tregs relative to conventional CD4+ T cells in tumors. Like in the in vitro model, the most statistically significant protein was Trps1.
Investigating the role of Trps1, the researchers performed CRISPR KO of the protein in hematopoietic lineages. In MCA205 tumor-bearing CHIME mice, Trps1 knockout induced spontaneous, durable tumor rejection in 7/13 mice and supported a significant survival advantage over controls. Importantly, there was no statistically significant decrease in the ex vivo suppressive capacity of Tregs containing Trps1-sgRNAs vs. control sgRNAs, and no consistent signs of autoimmunity or immunopathology were observed in peripheral tissues, suggesting that Trps1 is specifically relevant in tumors, where it contributes to the acquisition and maintenance of a more suppressive Treg phenotype.
Given the validation of Trps1 as an important MR in TI-Tregs, Obradovic, Ager, and Turunen et al. investigated the potential for therapeutic targeting of TI-Treg pathways by performing a systemic drug screen in which patient-derived, ex vivo-expanded TI-Tregs were treated with a library of 1,554 FDA-approved or investigational compounds. From 195 bioactive compounds that inhibited Treg viability by over 60%, the researchers selected those with the lowest maximum sublethal doses at 48 hours and evaluated the perturbation profiles of each drug at those doses. Based on inhibition of the 17-MR Treg signature, in vitro depletion of TI-Tregs vs. P-Tregs, OncoTreat predictions, the consistency of dose-dependent effects, and a number of other factors, the researchers identified gemcitabine as the top drug candidate for targeting TRPS1 and inhibiting TI-Tregs.
Further investigating the immunomodulatory effects of gemcitabine, the researchers found that in mice with late-stage MC38 tumors (resistant to anti-PD-1), low-dose gemcitabine (about 1/10 of the clinically relevant dose for mice) temporarily controlled MC38 progression and extended survival. Further, low-dose gemcitabine sensitized mice to anti-PD-1, with the combination inducing complete responses in 50% of mice and further extending survival over monotherapy. This effect was at least partly immune-mediated, as low-dose gemcitabine showed an advantage in immunocompetent mice vs. in NSG mice that, while modest, was sufficient to sensitize mice to anti-PD-1 and achieve complete responses and enhanced survival. At higher doses, the differential effects of gemcitabine in immunocompetent vs. NSG mice were neutralized.
In MC38-bearing mice, Tregs were the only subset to be reduced in absolute number in tumors after gemcitabine treatment, while splenic Tregs were not reduced. This effect was not due to reduced Treg proliferation in tumors, but rather due to inhibition of a highly suppressive Treg subset characterized by expression of Helios and CD103. Cluster analysis based on protein activity revealed a cluster that was highly enriched for the previously identified human TI-Treg MRs, including TRPS1. This cluster also had the highest expression of Helios, was more prevalent in tumors than in spleens, and was strongly reduced upon treatment with gemcitabine, suggesting that gemcitabine targets TI-Treg MR activity.
Overall, Obradovic, Ager, and Turunen et al. successfully uncovered and validated an MR signature that distinguished TI-Tregs from P-Tregs, and identified gemcitabine as a drug that at low (clinically sub-relevant) doses, could specifically target TI-Tregs and sensitize mice to immunotherapy – a finding that could prove useful in designing rational combination therapies in the clinic. The generalizable nature of this approach may also allow for the identification and targeting of MR proteins in other immunosuppressive subpopulations in the future.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, lead author Andrea Califano answered our questions.

What was the most surprising finding of this study for you?
The most surprising result was that modulating a single master regulator controlling the transcriptional state of tumor-resident regulatory T cells was sufficient to induce spontaneous remission in more than half of the tumors, even though biallelic silencing had an efficiency of less than 50%.
What is the outlook?
The fact that we were able to identify a drug (gemcitabine) which, at a dosage of 1/10th of the clinically relevant one, can inhibit the activity of the entire complement of master regulator (MR) proteins controlling the state of tumor resident Tregs is very promising because (a) this could be immediately tested in a clinical trial and (b) it may lead to identification and even design of additional drugs that may achieve the same result with even less toxicity.
What was the coolest thing you’ve learned (about) recently outside of work?
I have learned that over 200 Cenotes in Mexico are connected by underground caves that extend for thousands of miles and are just amazing to visit.



