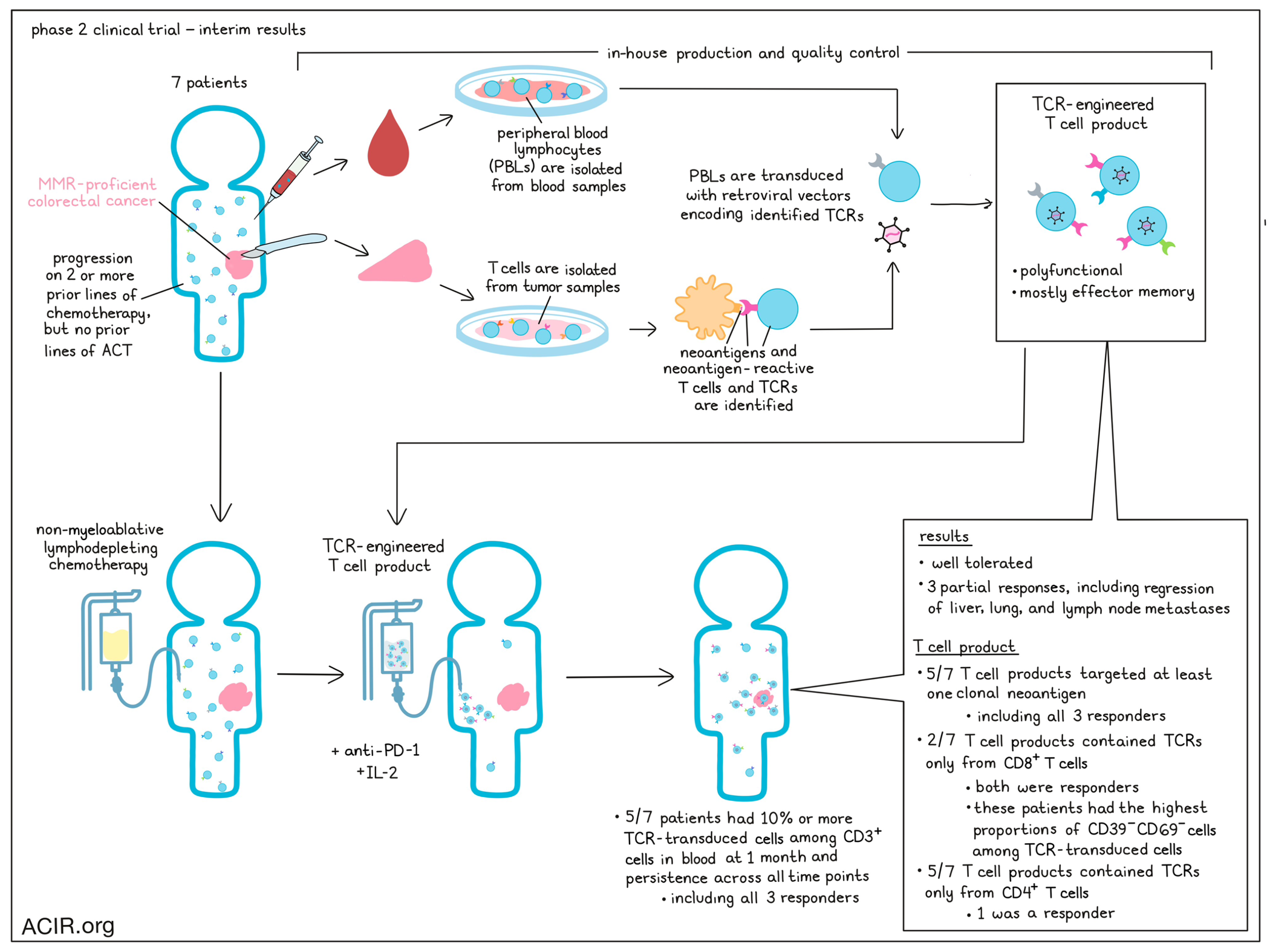
Targeting neoantigens using adoptive cell therapy (ACT) has been a promising approach to personalized cancer therapy, but isolating, identifying, and expanding relevant and high-quality T cells remains a challenge. In ongoing efforts to improve neoantigen-targeted ACT, Parkhurst et al. is conducting an ongoing phase 2 single-arm trial of TCR-engineered autologous peripheral blood lymphocytes (PBLs) in patients with various metastatic cancers. In a recent interim analysis, published in Nature Medicine, they report results from 7 patients with mismatch repair-proficient colorectal cancer, presenting the first evidence of clinical efficacy for personalized neoantigen-targeting TCR-engineered PBLs.
For this trial, Parkhurst et al. treated 7 patients with mismatch repair-proficient colorectal cancers that had progressed on two or more lines of chemotherapy, but were naive to treatment with ACT. Patients underwent partial surgical resections, and tumor samples were used to identify neoantigens and neoantigen-reactive T cells. Identified neoantigen-reactive TCRs were then encoded in retroviral vectors to be transduced into patient PBLs, under the hypothesis that PBLs would be less differentiated and less exhausted than tumor-infiltrating lymphocytes (TILs), enabling stronger antitumor immunity. Prior to ACT, patients underwent non-myeloablative lymphodepleting chemotherapy. Patients received a single infusion of TCR-engineered autologous PBLs, along with 4 doses of pembrolizumab, and cytokine support with IL-2 (most with high-dose IL-2). Overall, 3 of the 7 patients experienced partial responses, including regression of metastases in the liver, lungs, and lymph nodes, lasting from 4 to 7 months. The median PFS was 4.6 months. One patient experienced a grade 4 CRS, which resolved with supportive care, and all patients developed grade 3/4 cytopenias, consistent with lymphodepletion. Overall, treatment was well tolerated.
To develop the T cell products that were administered to patients in this trial, Parkhurst et al. utilized two different approaches to identify neoantigen-reactive TCRs. In the first approach, expanded T cells from samples of metastatic lesions were directly screened against tandem minigene constructs. These constructs could then be deconvoluted to identify the specific neoantigens. T cells that were activated against DCs loaded with the specific neoantigens could then be sorted, and the dominant TCR could be identified. Finally, these TCRs were encoded into retroviral vectors and transduced into PBLs, resulting in TCR-engineered PBLs that were reactive to the corresponding tumor neoantigens. Neoantigen binding to relevant HLAs was also predicted. In the second approach, single-cell digests were prepared from resected tumors, and individual T cells expressing CD8, PD-1, CD39, and TIGIT were sorted and sequenced to identify potential TCR α and β chains. Retroviral vectors encoding these TCRs were then transduced into PBLs and screened for neoantigen reactivity. All patients ultimately received T cell products that were at least 50% TCR-transduced.
Evaluating features of the T cell products as they might relate to responses, Parkhurst et al. noted that only 2 out of 7 patients received TCRs derived exclusively from CD8+ T cells, and both of these patients exhibited responses to therapy. Meanwhile, the remaining 5 out of the 7 patients received TCRs derived exclusively from CD4+ T cells, with only 1 of the patients responding to therapy, suggesting that the inclusion of CD8+ T cell-derived TCRs may more effectively contribute to responses. Further, in 5 of the 7 patients, at least one targeted neoantigen appeared to be clonal; all 3 patients who responded to treatment were among these 5, while neither of the patients who received TCRs targeting only subclonal mutations had a response.
A major distinction between this clinical trial and similar TCR-engineered T cell trials is the use of retroviral products generated from transiently transfected 293GP cells, which allows for small batches of retroviral products that can be produced and tested for quality control in-house. The T cell engineering process involved multiple rounds of stimulation with IL-2 and anti-CD3, and for some patients, depletion of either CD4+ or CD8+ T cells to enrich cells with the opposite coreceptor. This resulted in T cell products with a majority of cells expressing an effector memory (CD45RO+CD62L-) phenotype, without expression of CD27 (which would indicate a less differentiated phenotype). Additionally, there was substantial patient-to-patient variability in the portion of transduced cells expressing a CD39-CD69- phenotype (11–76%), which has previously been associated with responses to TIL therapies. The two patients who received CD8+ T cell-derived TCRs and responded to treatment had the highest percentages CD39-CD69- cells, suggesting a potential biomarker for efficacy in pre-treatment infusion products.
Evaluating the functionality of T cell products, the researchers found that T cell products for every patient were polyfunctional, secreting IFNγ, GM-CSF, IL-2, and granzyme B in response to mutant peptides. Production of other cytokines, chemokines, and perforin varied, but were not correlated with clinical responses.
In addition to evaluating T cell products prior to infusion, Parkhurst et al. evaluated T cells after infusion. At peak expansion, TCR-transduced T cells made up anywhere between 42 and 84% of T cells in the blood of patients. In 5 out of 7 patients, TCR-transduced cells could be detected at levels over 10% among CD3+ T cells at 1 month, and could be detected in the blood at every time point for which PBMC samples were available. These cells remained functional in ex vivo IFNγ secretion assays. The 5 patients with persistent T cell responses included all 3 responders. In one of the responding patients, transduced cells still made up about 20% of CD3+ cells in the blood more than 2 years after treatment. The phenotypes of persistent TCR-transduced T cells, ex vivo IFNγ responses, Tregs, serum IFNγ, and serum IL-2 were all uncorrelated with response. One patient developed antibodies to the transduced TCR, but this occurred late, and could not be directly linked to the patient’s lack of response.
Finally, Parkhurst et al. reported on 6 additional patients who were treated in an earlier iteration of a personalized TCR T cell trial, who had previously failed to respond to ACT therapy with TILs. While the researchers had hoped that treatment with a better quality T cell product consisting of a higher number of neoantigen-reactive T cells with less differentiated phenotypes might convert some patients, no responses were observed in this second-line ACT setting, possibly due to targeting of the same neoantigens that were ineffectively targeted previously.
These interim results suggest that personalized neoantigen-targeting TCR-tranduced PBLs have the potential to yield clinical responses, providing a proof of principle for this type of first-line ACT. They also begin to suggest that TCRs derived from CD8+ T cells, TCRs targeting clonal neoantigens, and increased CD39-CD69- transduced cells might be beneficial in the T cell infusion product, though more data is needed. This trial is currently ongoing, with additional results to follow.
Write-up and image by Lauren Hitchings



