Neoantigen vaccination and immune checkpoint blockade (ICB) are both strategies that can be employed to enhance antitumor T cell responses, but both are often met with resistance, and their use in combination has not been fully explored. Investigating these strategies in tumors with low mutation burdens, Dolina et al. found that immunization with neoantigens that are naturally recognized by both CD4+ and CD8+ T cells could induce antitumor immunity and overcome resistance to ICB. Interestingly, the CD4+ target antigen did not need to be tumor-specific, so long as it was linked to a tumor-specific CD8+ target antigen. The results of this study were recently published in The Journal of Clinical Investigation.
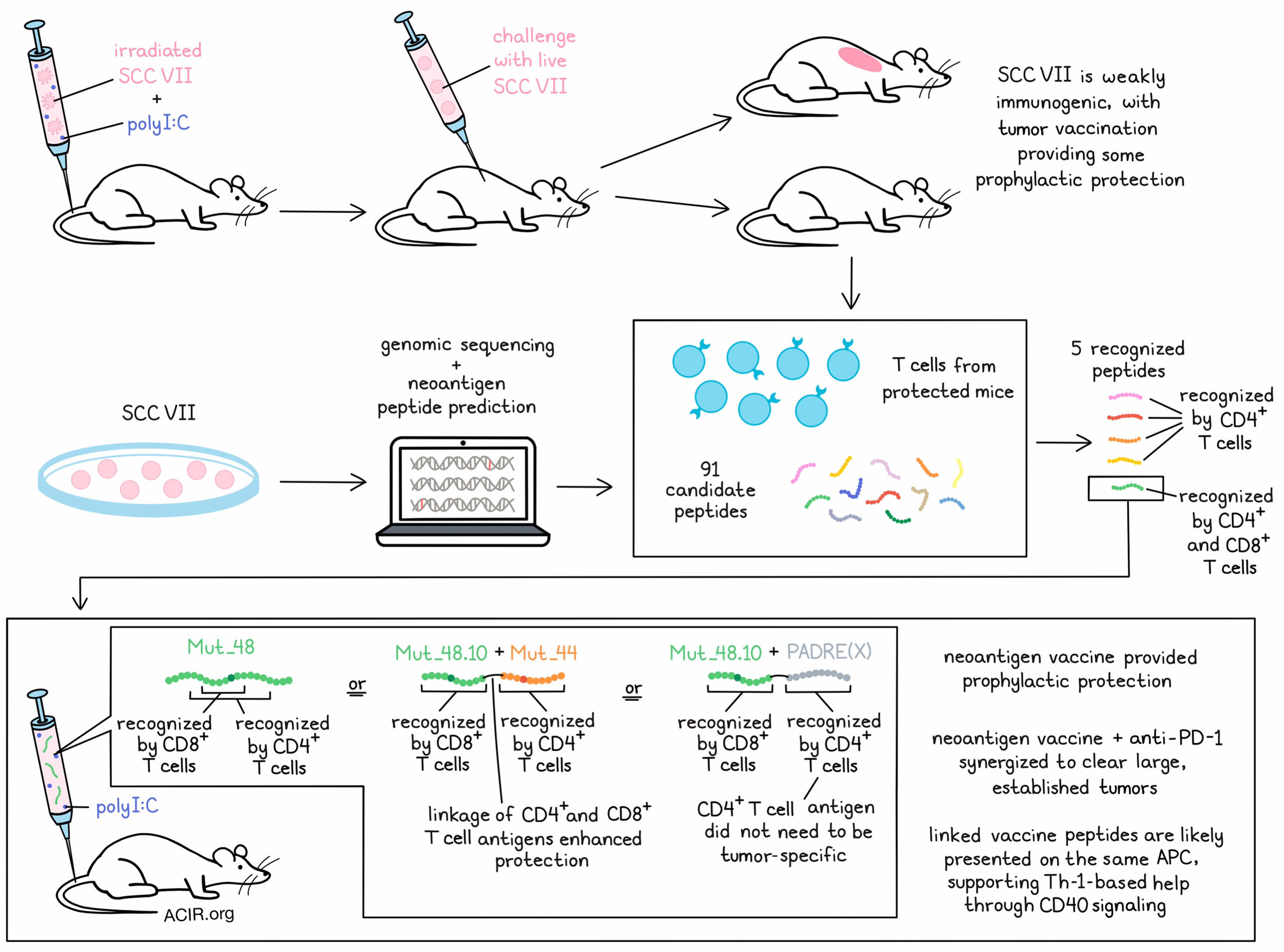
For their studies, Dolina et al. selected squamous cell carcinoma VII (SCC VII) – a spontaneously arising MHC-II- murine tumor that resembles human HNSCC and resists both chemo- and immunotherapy – as their tumor model. This model also contains tumor-initiating cancer stem cells (tCSC), which are marked by high CD44 and PD-L1 expression, are inherently resistant to treatments, and contribute to tumor heterogeneity. Immunizing mice with irradiated SCC VII prior to challenge with live tumor did not induce antitumor immunity, though adding polyI:C to the immunization led to rejection of the challenge, dependent on CD4+ and CD8+ T cells, suggesting that this model is naturally weakly immunogenic.
To detect neoantigens in SCC VII, the researchers used genomic sequencing and functional analysis. From the 39 mutations that were found to reach their expression threshold, the researchers generated 91 candidate peptides and tested them as targets for T cells by using them to restimulate mononuclear cells isolated from mice that had resisted a tumor challenge. Ultimately, the researchers were able to identify four mutated genes (including driver mutations) that produced five mutant peptides recognized by natural T cell responses. Immunization with a prime and boost containing a combination of these 5 peptides + polyI:C protected mice from challenge with SCC VII. Further, one of the peptides alone, MUT_48 + polyI:C was found to be sufficient to protect mice from tumor challenge.
Assessing the responses to these neoantigens, the researchers found that Mut_48 alone was recognized by both CD4+ and CD8+ T cells, while the others were recognized only by CD4+ T cells. Some CD8+ T cells also recognized two additional peptides, Mut_72 and Mut_73; however, these peptides were not capable of conferring protective immunity in vivo.
Digging deeper into the specific epitopes of MUT_48 that were recognized by CD4+ and CD8+ T cells, the researchers developed a panel of 10-mer and 15-mer peptides containing the MUT_48 H129Q mutation. CD8+ T cells identified the MUT_48.10 10-mer and CD8+ T cell minimal epitope (presented on H-2Kk). While CD4+ T cells did not recognize the MUT_48.10 10-mer, they did recognize the Mut_48.5 15-mer (presented on I-Ak), which contained the Mut_48.10 10-mer sequence. In vitro, the Mut_48.5 15-mer could elicit IFNγ responses from both CD4+ and CD8+ T cells. Similarly, in vivo, immunization with the Mut_48.5 15-mer was as protective against SCC VII as the full Mut_48 20-mer, while immunization with Mut_48.10 (recognized only by CD8+ T cells) was only partially protective, suggesting that CD4+ T cells were required for full prophylactic immunity.
To determine whether CD4+ T cell help needed to be tumor-specific, mice were immunized with the Mut_48.10 CD8+ T cell minimal epitope plus either SCC VII-derived Mut_44 (recognized by CD4+ T cells only) or with the PAN-DR epitope, PADRE(X), which is universally immunogenic to CD4+ T cells in Black6 mice. Both of these strategies were effective, eliciting a similar degree of protection as MUT_48 against SCC VII, suggesting that the CD4+ T cell epitope does not need to be tumor-specific. Further, covalent linkage of CD4+ and CD8+ T cell antigens enhanced protection compared to untethered peptides, suggesting that the enhanced protection from dually recognized peptides like MUT_48 likely stems from presentation of both the CD8+ target antigen and CD4+ helper antigen by the same APC, consistent with Th-mediated licensing of APCs to induce enhanced CD8+ T cell responses.
Next, Dolina et al. evaluated prophylactic neoantigen vaccination followed by ICB. When mice were treated prophylactically with the 5-peptide neoantigen vaccine alone, about half of mice experienced a late-phase relapse with SCC VII. However, with the addition of anti-PD-1 shortly after tumor implantation, these relapses could be prevented. Anti-PD-1 also expedited the elimination of tumors, increased memory T cell responses at day 42, and supported epitope spreading to Mut_72 and Mut_73 – targets that were not included in the vaccine. Similar results were not observed with anti-CTLA-4 delivered shortly after implantation.
When the combination of neoantigen vaccination and anti-PD-1 was tested against large, established tumors, they again showed synergy, inducing complete and durable tumor clearance that could not be achieved with either treatment alone. In this setting, combination treatment was found to increase CD8+ T cells among TIL and enhance Mut_48-specific memory T cell responses. Further, it appeared to inhibit metastasis to regional LNs.
Investigating the phenotypes of responding T cells in this setting, the researchers found that Mut_48 vaccination reduced naive and certain effector memory CD4+ and CD8+ T cell populations (in line with priming), while anti-PD-1 subtly expanded existing populations in TIL, including precursor/progenitor T cells, intermediate/exhausted cells, and an effector memory population of follicular T helper cells. Conventional CD4+ TILs were not cytotoxic, fitting with their role as helpers in this setting. Further, the addition of anti-PD-1 abrogated the expansion of terminally exhausted cells that was induced by vaccination alone, instead steering populations towards less differentiated states. Combination treatment also expanded a small population of precursor/progenitor T cells in the spleen and tumor-draining lymph nodes.
While CD4+ T cells were required for full efficacy in this model, CD8+ T cells were the critical effectors. Suspecting that CD4+ T cells provided Th-1-based help through CD40-mediated signaling in APCs, the researchers showed that an agonistic anti-CD40 cross-linking could replace CD4+ T cell help to support CD8+ T cell-mediated tumor eradication.
Finally, looking at tCSCs, Dolina et al. showed that while neither Mut_48 vaccination nor PD-1 blockade individually could significantly increase killing of SCC VII CD44lo tumor cells or CD44hi tCSC, combination treatment could effectively target both subsets, suggesting that this strategy can overcome resistance mechanisms.
Overall, these results show that vaccinations with neoantigens naturally recognized by the immune system are more effective when peptides simultaneously stimulate helper CD4+ and effector CD8+ T cells, likely via the same APC. Further, neoantigen vaccination can synergize with anti-PD-1 to induce both prophylactic and therapeutic antitumor immunity, which could be translationally relevant.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Joseph Dolina answered our questions.
What was the most surprising finding of this study for you?
When scanning peptide reactivity for individual CD4+ and CD8+ epitopes, we were surprised that full protection from tumor burden was solely elicited by a neoantigen that concurrently stimulated both T cell subsets. Neither CD4+ nor CD8+ T cell neoantigen vaccination alone was sufficient to cure mice of cancer, and this made sense! This is how CD4+ T cell help works… synchronized loading of MHC I and II epitopes on the surface of the same antigen-presenting cell. Further, helped tumor-specific CD8+ T cells were more capable of forming memory and resisting exhaustion.
What is the outlook?
This work has immediate impacts into vaccine design for clinical trials – establishing the need to include verified CD4+ T cell neoepitopes for supporting stable antitumor CD8+ T cell responses. However, more research is needed to understand how peptide affinity and the quality of CD4+ T cell help affects CD8+ T cell differentiation in the periphery and once inside the harsh tumor microenvironment. Function-based methods for altering tumor vaccination behavior, as outlined in this work, can also be used to train neoantigen prediction algorithms to better model how T cells respond to tumor neoantigens.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently became a parent to Joseph the 5th (yes there is a lineage of us) during the writing of this manuscript. He has opened my world and touched my heart to levels I never could have expected.



