PD-1 axis blockade has been proven time and again to be a successful immunotherapy for a wide variety of cancer types, and it is assumed to work by reversing or preventing T cell exhaustion. However, the exact way by which this cancer immunotherapy works has not been fully elucidated, and recent studies have shown that the mechanism behind PD-1 axis blockade may be more complex than previously thought. Armed with the knowledge that intratumoral myeloid cells express PD-L1 on their surface, Oh et al. set out to explore the role of different myeloid cell populations in the regulation of T cell immunity and PD-1/PD-L1 blockade. The results were recently published in Nature Cancer.
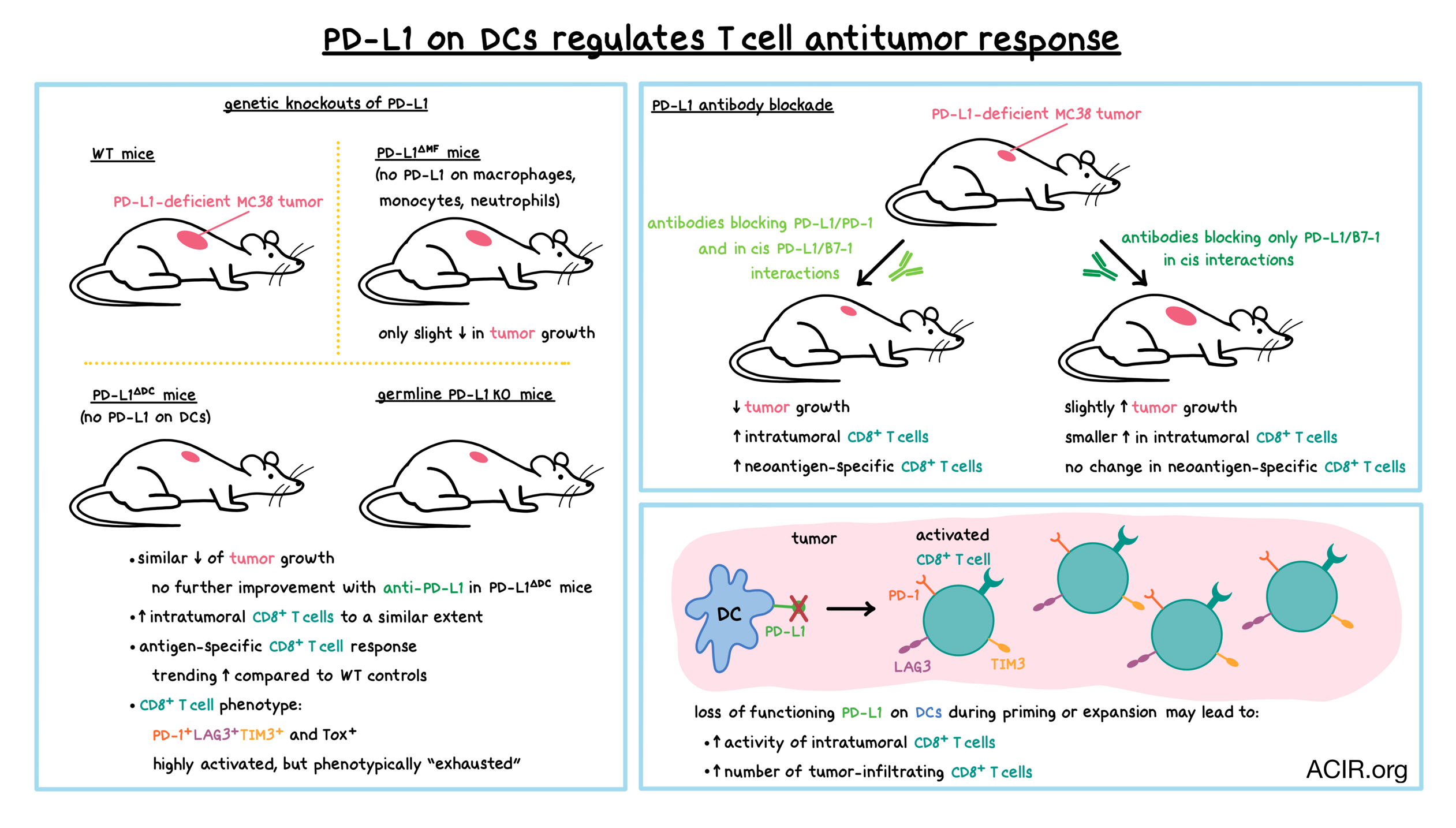
Knowing that PD-L1 and B7-1 (a CD28 ligand) interact with each other in cis on the surface of myeloid cells, Oh et al. began by exploring how disrupting this interaction would affect the endogenous antitumor response. To initially investigate a model system, the researchers generated Jurkat cells with chimeric receptors comprising an extracellular PD-1 domain, an intracellular human CD3ζ signaling chain, and a luciferase reporter gene. K562 cells expressing PD-L1, B7-1, or both were used as artificial APCs. After confirming that binding in cis between PD-L1 and B7-1 reduced PD-1 signaling, but not CD28 signaling, the researchers created anti-PD-L1 antibodies that selectively inhibited interactions between PD-L1 and PD-1, PD-L1 and B7-1, or both. As expected, selectively disrupting the PD-L1/B7-1 interactions made more PD-L1 available, and signaling increased in this assay, while blocking PD-L1/PD-1 interactions had the opposite effect.
Consistent with in vitro observations, in mice with PD-L1-deficient MC38 colorectal tumors where only host cells were the source of PD-L1, antibody blockade of both PD-1/PD-L1 and PD-L1/B7-1 interactions reduced tumor growth, while blocking only PD-L1/B7-1 interactions slightly accelerated tumor growth due to an increased availability of PD-L1. Blocking both interactions increased the number of tumor-infiltrating CD8+ T cells, including CD8+ T cells specific for the tumor neoantigen M86. In contrast, blocking only the PD-L1/B7-1 interactions led to a smaller increase in CD8+ T cells, and no increase in M86-specific CD8+ T cells. Together, these experiments showed that PD-L1 expression on antigen-presenting cells (APCs) played a role in antitumor T cell responses via PD-1 interactions.
Analysis of the tumor-infiltrating myeloid populations at days 3, 7, and 14 post tumor inoculation revealed that the majority of myeloid cells were macrophages, which far outnumbered cross-presenting dendritic cells (DCs). Most myeloid cells within tumors and tumor-draining lymph nodes expressed PD-L1 without co-expressing B7-1 (but still expressing B7-2, which does not interact in cis with PD-L1), thus leaving plenty of PD-L1 free to interact with PD-1 on T cells. Most intratumoral PD-L1+ myeloid cells were macrophages (~75%), while cross-presenting DCs accounted for less than 2% of this population by day 14 post tumor inoculation. A similar PD-L1 expression distribution was observed in RNASeq data from human non-small cell lung cancer (NSCLC) tumors. Interestingly, however, mature DCs in NSCLC tumors had the highest level of PD-L1 (and B7-1) expression on a per cell basis among intratumoral myeloid populations.
To determine which PD-L1-expressing myeloid cells are the primary regulators of antitumor immunity, Oh et al. created mice with DC-specific PD-L1 deficiency (PD-L1ΔDC mice). PD-L1 loss was greatest in the CD11b-F4/80- DC subset. The researchers also created PD-L1ΔMF mice, in which PD-L1 was deleted in macrophages, monocytes, and neutrophils, while DCs were largely unaffected. In animals bearing PD-L1-deficient MC38 tumors, mice with total PD-L1 knockout (KO) controlled tumor growth better than wild-type (WT) mice, as observed previously. PD-L1ΔDC mice controlled tumors as effectively as PD-L1 KO mice, while PD-L1ΔMF mice were not effective, reducing tumor growth only slightly compared to WT controls. Similar results were observed in mice bearing PD-L1-sufficient tumors. Treating tumor-bearing PD-L1ΔDC mice with anti-PD-L1 antibodies that blocked either only the PD-L1/B7-1 interactions or both the PD-L1/B7-1 interactions and PD-L1/PD-1 interactions did not further enhance tumor control. Together, these results indicated that despite their lower frequency, DCs played a bigger, and crucial role as a source of PD-L1 in PD-L1/PD-1 regulation of the antitumor response compared with the more abundant macrophages. Furthermore, PD-L1/B7-1 interactions in cis on DCs, but not on macrophages, played a role in limiting PD-1 signaling and in activating T cells.
Analysis of T cell responses at day 10 post inoculation revealed increased frequency and number of CD8+ T cells within PD-L1-deficient MC38 tumors of PD-L1 KO and PD-L1ΔDC mice compared to WT controls. Levels of intratumoral CD8+ T cells increased to a similar extent in both types of mice. Antigen-specific CD8+ T cell responses were variable, but also trended upward in PD-L1 KO and PD-L1ΔDC mice (a trend separately confirmed measuring vaccine-induced SIINFEKL responses), further highlighting the important role of DC PD-L1 in regulating the immune response. In all three types of mice, CD8+ T cells exhibited a PD-1+LAG3+TIM3+ phenotype, typically consistent with exhaustion.
By day 23 post tumor inoculation, tumors were much smaller in PD-L1-deficient (PD-L1 KO and PD-L1ΔDC) mice than in WT controls. At that time point, neoantigen-specific, tumor-infiltrating CD8+ T cells were observed in all three types of mice, as were highly activated PD-1+LAG3+TIM3+ CD8+ T cells (the latter were increased in PD-L1 KO and PD-L1ΔDC mice compared to controls). PD-1+Tox+ CD8+ T cells were also increased in PD-L1 KO and PD-L1ΔDC mice. Together, these results demonstrated that in PD-L1 KO and PD-L1ΔDC mice compared to controls, intratumoral CD8+ T cells were highly activated. Surprisingly, given the improved tumor control, the CD8+ T cells were also more likely to appear “exhausted”, challenging the notion that the phenotypic state of exhaustion necessarily reduces antitumor activity. The researchers hypothesized that the loss of PD-L1 on DCs during priming or expansion may lead to enhanced activity of the exhausted intratumoral CD8+ T cells or may result in increasing the number of tumor-infiltrating CD8+ T cells, or it may act in both ways. Additionally, these experiments clarified that PD-L1 was not required to induce an exhausted phenotype in intratumoral T cells.
Overall, Oh et al. demonstrated that among intratumoral myeloid populations, PD-L1-expressing DCs play a crucial role in ultimately regulating antitumor T cell responses via PD-L1/B7-1 interactions in cis on DCs and PD-L1/PD-1 interactions between DCs and T cells. These results suggest that PD-1/PD-L1 blockade may work in a more complex fashion than previously thought: in addition to reversing or preventing T cell exhaustion, which unexpectedly also depends on DC/T cell interactions, PD-1 axis blockade may play a role during T cell priming and expansion. A more complete understanding of these mechanisms will be required to optimize anti-PD-1/anti-PD-L1 cancer immunotherapy.
by Anna Scherer
Meet the researcher
This week, first author Soyoung Oh answered our questions.

What prompted you to tackle this research question?
Blockade of PD-1 or PD-L1 has long been assumed to function by restoring the functionality of exhausted tumor-infiltrating CD8+ T cells. This paradigm centers the mechanism of PD-1/PD-L1 blockade in the tumor, and although PD-L1 can be expressed by both tumor cells and immune cells, the prevailing view is that blocking PD-L1 expressed by tumor cells drives the efficacy of these treatments. However, we were intrigued by several observations indicating a more complex picture than one in which PD-L1 on tumor cells is the primary target of anti-PD-L1 therapies. Studies in mice had shown that tumoral and non-tumoral (e.g. immune cell) sources of PD-L1 play non-redundant roles in regulating antitumor immunity. In human cancers, PD-L1 can be expressed largely by tumor-infiltrating immune cells, which also serves as a predictor of response to therapy. We wanted to determine whether a specific (non-tumoral) cellular source of PD-L1 might have a dominant effect on the regulation of antitumor T cell responses. Although PD-L1 can be expressed by multiple types of immune cells, we were most interested in the contribution of myeloid cells, and in particular in the role of PD-L1 on dendritic cells. Notably, myeloid cells (but not tumor cells) can also express B7-1, a ligand for the costimulatory CD28 pathway that we had recently found was inhibited by PD-1 signaling.
What was the most surprising finding of this study for you?
While I suspected that dendritic cells would be an important source of PD-L1, it was amazing to see that mice with dendritic cell-specific deletion of PD-L1 controlled tumors as effectively as mice with global deficiency of PD-L1. This was even more striking since mice with macrophage-specific deletion of PD-L1 were not as effective as either the DC-specific or global PD-L1 knockout animals in controlling tumors.
What was the coolest thing you’ve learned (about) recently outside of work?
My five-year-old son is obsessed with fact books about animals. Thanks to reading with him, I recently learned about the “immortal jellyfish”, Turritopsis dohrnii. In response to certain conditions, this species can transform from its mature, adult state (medusa) into an immature, less differentiated state (polyp). Incredibly, there seems to be no limit to the number of times Turritopsis dohrnii can undergo this process of transdifferentiation



