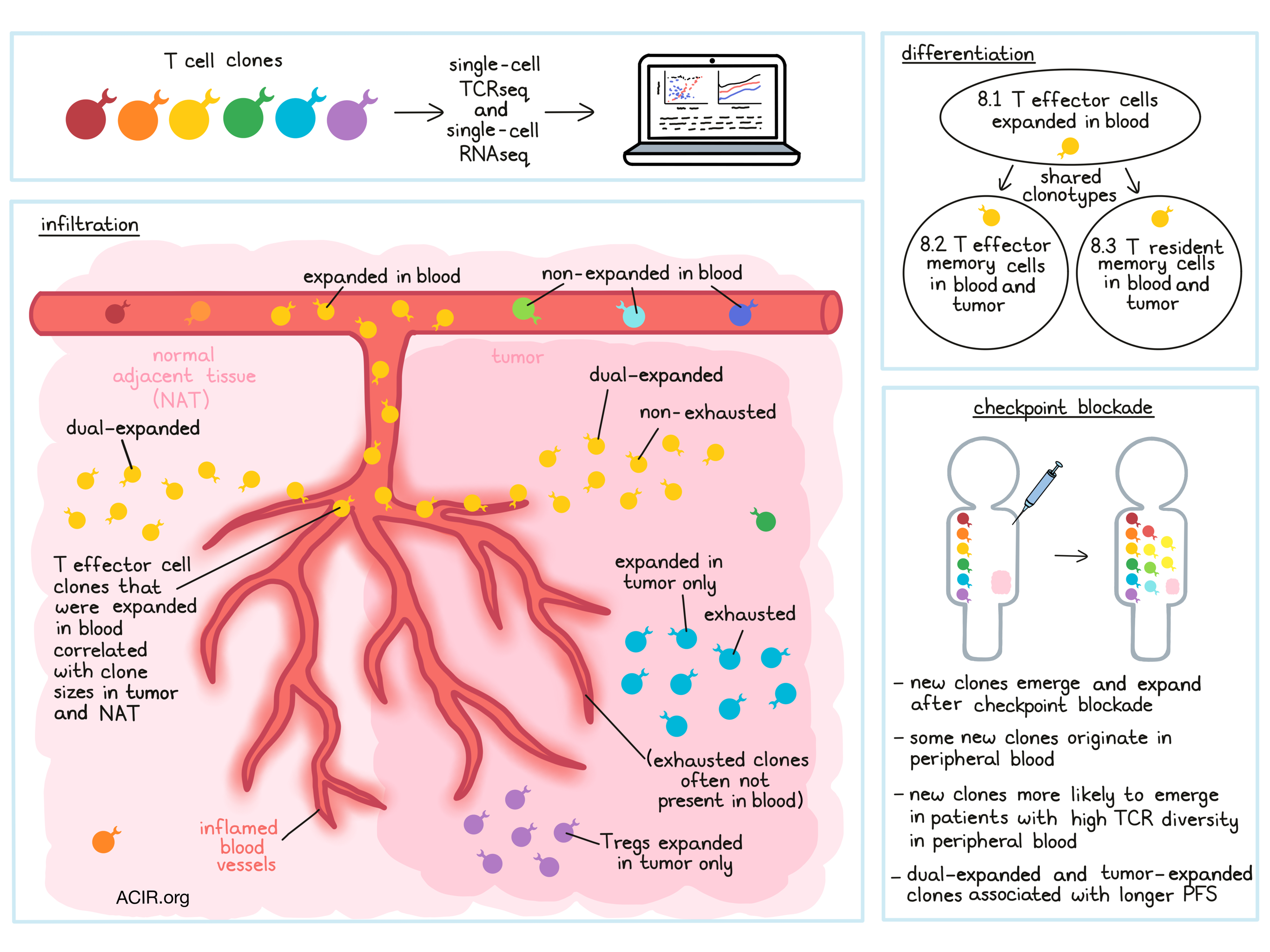
In an effort to investigate the origin, behavior, and fate of T cells in patients with cancer, Wu et al. sequenced 330 million mRNA transcripts from 141,623 T cells, which had been isolated from the tumors and normal adjacent tissue (NAT) of 14 treatment-naive patients with 4 different types of cancer; peripheral blood samples were available for 4 patients. Further, single-cell TCR sequencing of cells identified 56,975 distinct clonotypes based on matching CDR3 regions, which allowed for analysis of clonal expansion and tracking of T cell lineages across tissues. The results of analysis of this data set, and several external data sets, were recently published in Nature.
Analysis of clonotypes in the 14 patients with lung (6), endometrial (3), renal (3), or colorectal (2) cancer showed that 9-18% of clonotypes were expanded (found more than once in a patient). To better describe the locations of T cell clones, Wu et al. defined T cell clones that are expanded in both the tumor and the NAT as dual-expanded. In some patients, clonal expansion appeared coordinated between the tumor and NAT, while in others, divergent patterns between the two compartments were observed, leading the researchers to hypothesize that parallel expansion in the tumor and the NAT may represent comparable infiltration of T cells from the periphery through inflamed blood vessels into both tissues. Evidence from matched blood samples supported this hypothesis, as highly expanded clones in the blood were frequently found to be dual-expanded. Further, an aggregate measure of overall peripheral expansion (fraction of peripheral clones that were expanded) was associated with tumor and tissue infiltration by blood-expanded clones, suggesting a strong relationship between peripheral clonal expansion and parallel infiltration into tumors and NAT.
Using scRNAseq data from individual T cells, Wu et al. grouped similar cells into transcriptomic clusters. Characterization of these clusters identified T effector cells, T effector memory, Tregs, and cells with resident memory-like characteristics (CD103+, Hobit+). Within the resident memory-like compartment, subclusters could be identified based on levels of PDCD1 expression and other factors. These subclusters showed increased prevalence in the tumor vs. NAT, supporting a relationship between PD-1 expression and exposure to antigens in the TME. Several of the clusters that were identified did not match published reference gene signatures, but showed evidence of exhaustion and/or tissue residency.
To better understand the relationship between clonal expansion and transcriptional behavior in T cells, Wu et al. performed a combined analysis using both scTCRseq and scRNAseq data. In this analysis, clones were found to exhibit a range of transcriptional diversity, with some clones falling homogeneously into a single cluster and others falling into multiple clusters. Clones that fell into multiple clusters, however, typically fell into a “primary” cluster, which could serve as an approximate phenotype for subsequent analysis. Three of the CD8+ T cell clusters – deemed 8.1 T effector, 8.2 T effector memory, and 8.3 T resident memory – were largely dual-expanded. Most CD4+ T cell clusters did not show evidence of clonal expansion, with a few exceptions, including clonally expanded Tregs in the tumor.
Mapping primary clusters onto clones from the four patients for whom blood samples were available, Wu et al. found that many clones exhibiting parallel dual expansion were 8.1 T effector clones, and that the presence of blood-expanded clones in cluster 8.1 significantly correlated with tumor and NAT clone sizes, suggesting a role in tumor infiltration. Interestingly, while clones in cluster 8.1 constituted only a small fraction of all clones, they had a major influence on T cell composition in blood and tumors. Investigating expansion patterns, the researchers observed that clones in clusters 8.2 and 8.3 that were expanded in blood and tumors frequently shared clonotypes with blood-expanded clones in cluster 8.1, suggesting that 8.1 T effector cells may also differentiate into memory T cell phenotypes.
To better understand the dynamics of clone sizes across tumors, NAT, and blood over the course of treatment with immunotherapy, the researchers analyzed an external dataset of pre- and post-treatment T cells from patients with basal cell carcinomas who were treated with an anti-PD-L1 drug. Comparing pre- and post-treatment samples, Wu et al. observed that new clones emerged after treatment, and that at least some of the new CD8+ T clones that emerged originated in peripheral blood. Individual patients differed in the number and expansion of new clones, though patients with a more diverse peripheral blood clonal repertoire (averaged pre- and post-treatment diversity) were more likely to see the emergence and expansion of new clones after treatment.
In the two patients with the greatest peripheral clonal diversity, the researchers found that non-exhausted clones were more likely to be blood-associated, while exhausted clones were often not present in the blood. Non-exhausted clones in the blood also had significantly greater clone sizes in tumors after treatment compared to clones not found in blood. Based on their observations, the researchers suggest that some clonal expansion can originate within tumors, generating exhausted T cells, while other expanded clones may originate in the periphery and introduce novel non-exhausted clones into the TME.
Given that clonal expansion of T cells in the periphery and in tumors was observed to be variable, the researchers hypothesized that this variability might impact PD-1/PD-L1 checkpoint blockade therapies. To this end, Wu et al. evaluated bulk RNAseq data for tumor samples taken across three phase II clinical trials of an anti-PD-L1 drug. Unsurprisingly, gene signatures of clonally expanded T cells correlated highly with CD8A expression, which is often considered a predictor of response to immunotherapy. However, CCL5 expression (a chemoattractant protein associated with T cell activation) showed a stronger association with progression-free survival (PFS) than CD8A expression.
Gene signatures for dual-expanded or tumor-expanded clones were associated with greater PFS. In general, high CD8A expression and gene signatures for dual expansion or tumor-specific T cell expansion coincided in patients, though a few patients showed high CD8A expression but low expansion signature genes. In these patients, treatment with anti-PD-1 was associated with shorter PFS compared to patients with both markers. Further, tumor-specific expansion and dual expansion had additive predictive power, suggesting that both locally and peripherally expanding T cells may contribute independently to clinical responses.
Overall, the extensive analyses by Wu et al. suggest that dual-expanded T cells derived from peripherally expanded clones may best represent the overall source of effective immune response. As it relates to checkpoint blockade, this model presents an alternative to the notion that checkpoint blockade reverses exhaustion or terminal differentiation in chronically stimulated T cells. Instead, this model suggests that checkpoint blockade may act by enhancing the supply and/or activity of non-exhausted T cells from the periphery. From a practical standpoint, this research also highlights the potential for using peripheral blood sampling to characterize clinically relevant T cells.
by Lauren Hitchings
To receive our next digest in your inbox, subscribe here!
Meet the researcher
This week, first author Thomas Wu and lead author Jane Grogan answered our questions.

What prompted you to tackle this research question?
TW: We did not initially consider the peripheral component of the immune system. We started with a general desire to understand the populations of T cells in tumors, and from our colleagues' work on the stroma surrounding tumors, we knew that it was important to obtain samples from adjacent regions as well. However, after studying our initial set of samples and discovering an unexpectedly large number of dual-expanded clones, we knew it was critical to obtain additional samples from patient blood, which led to our main findings regarding peripheral T cell expansion and tumor infiltration. We also realized that these phenomena could explain a number of recent observations by others, such as the presence of T cells reactive against non-tumor antigens and clonal replacement in tumors. The variability of peripheral T cell expansion across patients also led us to consider its possible relationship with clinical response.
JG: Over the past several years, a great deal has been learned from both clinical and preclinical investigations concerning the cycle of events that must occur to generate effective T cell immunity in cancer. These include the presentation of tumor-associated antigens (often patient-specific mutant neoantigens) by dendritic cells to T cells in tumor-draining lymph nodes, the homing of antigen-specific T cells to the tumor site, and the recognition and killing of cancer cell targets. It is also well known that in the tumor bed, tumor-infiltrating T lymphocytes often exhibit a terminally differentiated or “exhausted” phenotype characterized by high expression of PD-1 and other negative regulators. PD-1, in this view, is activated to induce exhaustion by binding high levels of PD-L1 expressed by many responsive tumors. Response to PD-L1/PD-1 blockade, therefore, is often thought to effect a reversal of this phenotype with the resulting “reinvigorated” T cells acting to kill their targets. However, recent observations have begun to question this model. The phenotype of terminally exhausted T cells appear to be epigenetically locked and difficult to alter. Indeed, PD-L1/PD-1 blockade causes the expansion of a stem-like T memory cell compartment, distinct from the exhausted cells. A major limitation to understanding the origin and fate of T cells in tumor immunity is the lack of quantitative information on the distribution of individual T cell clonotypes in cancer patients, which we sought to collect and examine in this study.
What was the most surprising finding of this study for you?
TW: We are surprised at how well our findings are corroborated by external datasets, even though they come from different sequencing technologies and different cancer types. The commonalities we found across datasets suggest that our results might further the development of blood-based TCR repertoires for diagnosis, treatment, triage, and monitoring in cancer immunotherapy.
JG: Analysis of our data (along with several external datasets) shows that intratumoral T cells, especially in responsive patients, are continuously replenished with fresh, non-exhausted replacement cells from sites outside of the tumor, suggesting continued activity of the cancer immunity cycle in these patients, the acceleration of which may be associated with clinical response. These observations suggest that the supply of T cells in tumors may depend on the availability of effector and effector memory T cells in the periphery. They also raise the possibility that, in some patients, sampling T cells in blood may provide information about the TCR repertoire of tissue-resident T cells, expanding the possibilities for “liquid biopsies”.
What was the coolest thing you’ve learned (about) recently outside of work?
TW: I have started swimming again after being away from the pool for a couple of years. I’m now using these goggles with a smart display that shows me my splits after each lap, and I’m finding how motivating it can be to have constant feedback.
JG: That Antarctica recently experienced its single hottest day ever recorded, hitting a high of 69.35 degrees Fahrenheit (20.75 degrees Celsius), and appreciating how scientists record the world around them in a way that can influence and improve all of our lives.



