
This week’s extensive special feature covers select talks from the 37th Annual Meeting of the Society for Immunotherapy of Cancer (SITC) in Boston, MA and virtually. We are proud to recognize Takeda for sponsorship of our coverage for this conference.
We have organized the content by topics below.
Keynote address
Padmanee Sharma
Big data
Hanna Carter
Nir Hacohen
Benjamin D. Greenbaum and Vinod Balachandran
Michael Birnbaum
Robert Schreiber
Cell therapies
Stefanie Mandl
Amod A. Sarnaik
Michael Ball
Checkpoint blockade
Stephanie Dougan
Siwen Hu-Lieskovan
Ira Mellman
Vijay Kuchroo
Antibody-based therapies
Dimitri Skokos
Jamie Spangler
Tumor microenvironment
Thomas Gajewski
Anthony Rongvaux
Mads Hald Andersen
In situ vaccination and cancer vaccines
Edward J. Gane
Thomas Marron
Oncolytic viruses
Igor Puzanov
Keynote address
From the Clinic to the Lab: Investigating Mechanisms of Response and Resistance to Immune Checkpoint Therapy - Padmanee Sharma, MD, PhD - The University of Texas MD Anderson Cancer Center
Padmanee Sharma highlighted how neoadjuvant (pre-surgical) clinical trials allow us to move into earlier stages of disease and obtain sufficient samples, which can provide insights into clinical signals, biomarkers, and mechanisms of action. For example, the first neoadjuvant clinical trial of anti-CTLA-4 in patients with bladder cancer, done in 2006 (prior to FDA approval), provided tissue samples collected at the time of surgery, which allowed for immune monitoring studies that helped to identify mechanisms of response and resistance. Samples obtained from surgical resections in another neoadjuvant trial of anti-CTLA-4 plus anti-PD-L1 in patients with localized bladder cancer revealed that the responders in the trial had tertiary lymphoid structures with increased ICOS-expressing CD4+ T cells. Interrogating the role of ICOS in mice, Sharma and colleagues showed that ICOS/ICOSL KO mice demonstrated impaired tumor control with anti-CTLA-4 therapy, whereas targeting ICOS plus anti-CTLA-4 improved antitumor responses against B16 melanoma. In a trial in lung cancer, anti-PD-1 and anti-CTLA-4 led to a number of major pathological responses (mPR). In a trial in cutaneous squamous cell carcinoma, anti-PD-1 neoadjuvant therapy led to the pathological responses. In yet another trial, neoadjuvant and adjuvant immune checkpoint blockade was evaluated in patients with high-risk, resectable metastatic melanoma. Here, anti-PD-1 monotherapy showed a pCR rate of 25%, while the combination of anti-PD-1 and anti-CTLA-4 showed a complete pathological response rate of 45%, demonstrating the potential benefit of combination therapy. Investigating “cold” versus “hot” tumor microenvironments, Sharma and colleagues performed a neoadjuvant therapy trial in 2009 in prostate cancer, which showed that anti-CTLA-4 converted cold tumors into hot, with increased immune cell infiltration. However, compensatory inhibitory pathways, such as VISTA and PD-L1 (expressed in CD68+ macrophages with mutual exclusivity) were upregulated in these tumors as well. Sharma also described how analysis of GBM samples led to identification of CD73 as a new target on myeloid cells in the GBM TME, and how their analysis of the epigenetic effects of EZH2 driving Foxp3 stabilization on regulatory T cells opens upstream elements as potential new targets. Finally, Sharma described a pilot safety trial in stage 4 metastatic clear cell renal cell carcinoma (ccRCC) in which immune checkpoint blockade prior to debulking surgery of a larger lesion (or lesions) provided good safety data. An ad hoc analysis suggested a potential clinical benefit, and tumor tissues from these patients showed that a higher IFNγ signature was associated with partial response at the population level, but not at the individual level. Tumors from patients with high IFNγ signatures, both responders and non-responders, showed that while a high IFNγ signature alone may not be sufficient to predict response, spatial interactions between T cell, B cells, and myeloid cells could predict better outcomes. Together, these results support the use of neoadjuvant clinical trials both to treat patients and to investigate emerging therapies and combinations.
Big data
Germline Immune Variants Influence Cancer Risk and Response to Immunotherapy - Hannah Carter, PhD – UC San Diego School of Medicine
Precision cancer medicine can be used to identify patients at risk of cancer for preventative measures and screening. But it can also be used for patient stratification for prognostic and treatment purposes. Hannah Carter presented research on germline immune variants and their role in cancer risk and response to immune checkpoint blockade. Initial research focused on MHC genotype. Driver mutations were found to be poorly presented by MHC, which may affect response to immune checkpoint blockade (ICB), as the therapy requires effective neoantigen presentation to cytotoxic T cells. In a study assessing 77 patients who had received ICB monotherapy, somatic mutations were profiled using the Foundation Medicine panel to assess driver mutations that were effectively presented. Although high tumor mutational burden (TMB) correlated with better prognosis, incorporating driver mutations presented on MHC as an additional criteria could separate responders and non-responders among patients with high TMB, suggesting that effective neoantigens presentation is required for response to ICB. More recent research focused on determining the influence of germline variants on immune traits, and how this affects immune infiltration and innate and adaptive responses. The researchers investigated molecular characteristics and germline associations with the tumor immune microenvironment (TIME). Of the assessed 741 immune markers, 140 had significant genetic components driving phenotypic variance, and overall, 890 associations were identified, with 858 unique variants. The phenotypes with most associations were ERAP1, ERAP2, CCBL2, and DHFR. Next, TIME associations from previous germline studies in the literature were used to detect cancer-relevant TIME associations. Using a colocalization analysis that can prioritize likely causal genes, most hits were found associated with risk, then survival and ICB response. TIME-SNPs were found to be associated with cancer risk using UK biobank data from melanoma and prostate cancer. Then, TIME-SNPs were analyzed in the context of immunotherapy response by pooling together a set of cohorts with exome sequencing data. When comparing responders to non-responders, the total burden of TIME-SNPs was predictive in melanoma, renal cell carcinoma, and non-small cell lung cancer datasets, suggesting TIME-SNPs were associated with ICB response. For one TIME-SNP, Cathepsin S (CTSS), a validation study was performed in the MC38 mouse model. CTSS is important for MHC-II antigen presentation, and high CTSS is associated with poor outcomes in both humans and mice. Mice were treated with anti-PD-1, a small molecule CTSS inhibitor, or the combination, and the most tumor growth reduction was seen in the group treated with the combination. The therapy remodeled the TIME by causing a shift in the macrophage population from the suppressive M2-like population to M1-like macrophages. Further analyses suggested that TIME-SNPs impact both innate and adaptive immunity. Most of the SNPs the group identified were associated with antigen processing, and there was also a clear link with the cytotoxic function of effector cells. Therefore, the inherited genome can influence host antitumor immunity, and studying SNP associations may provide new targets for therapy.
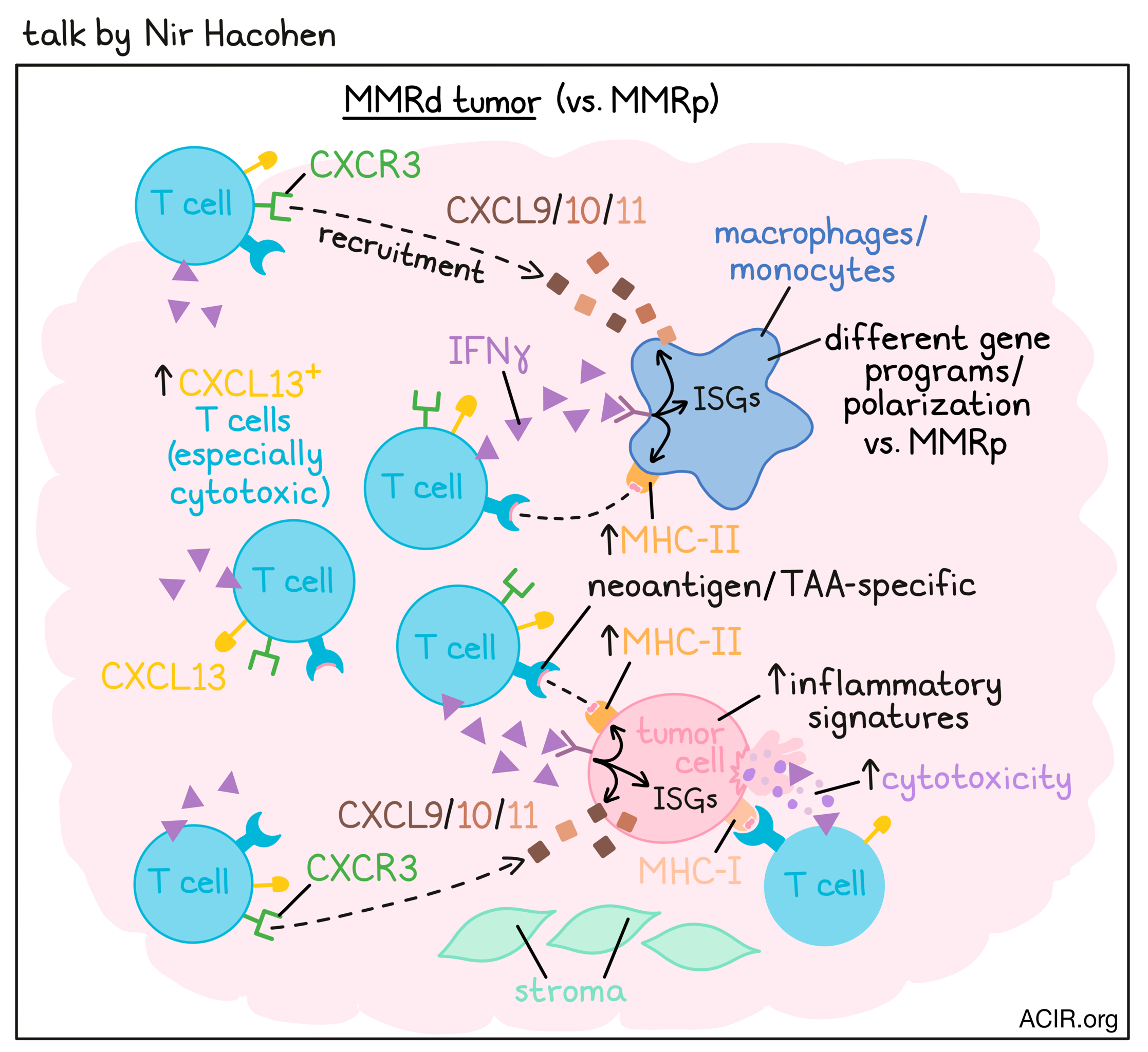
Discovering the Rules of Human Tumor Immunity - Nir Hacohen, PhD - Massachusetts General Research Institute
Shining light on the question how immune responses are coordinated in tumors, Nir Hacohen asked whether there is a spatial organization of immune responses in tumors, and what mechanisms might underlie the dramatic difference in response of mismatch repair-deficient (MMRd) versus MMR-proficient (MMRp) tumors to immunotherapy. Analyzing scRNAseq data from ~400,000 cells across colorectal cancer (CRC) and adjacent normal colon tissue, Hacohen and team identified 87 clusters across 7 cell lineages. In the T cell compartment, especially cytotoxic CXCL13+ T cells were enriched in MMRd tumors compared to MMRp CRC and normal tissue. CXCL13+ T cells were present in close proximity to cancer cells, but not in tertiary lymphoid structures. Further, CXCL13 served as a marker for neoantigen- and tumor antigen-reactive T cells in patients. In the myeloid compartment, Hacohen found that monocytes and macrophages acquired tumor-specific gene programs and different polarization states in MMRd versus MMRp CRC. Gene programs in malignant cells were also altered in MMRd versus MMRp CRC, with higher inflammatory and interferon-stimulated gene (ISG)/MHC-II signatures in MMRd CRC. Hacohen used co-variation of program activities across patient specimens to predict multicellular interaction networks (hubs) and identified 7 hubs in MMRd and 9 in MMRp tumors. A putative antitumor immunity hub in MMRd CRC contained the CXCL13 T cell program, the malignant cell ISG/MHC program, and an ISG and DC program in myeloid cells. This hub showed higher correlation, activity, and connectivity in MMRd versus MMRp CRC. In a positive feedback loop in this hub, IFNγ from activated CXCL13+ T cells induced ISG programs in malignant and myeloid cells, and CXCR3 ligands (CXCL9/10/11) from malignant and myeloid cells recruited more CXCR3+CXCL13+ T cells. FISH microscopy images demonstrated that CXCL10/11+ malignant cells associated with CXCL13+IFNγ+ T cells were organized into focal hubs at the tumor–stromal interface. In an attempt to further enhance these antitumor immunity hubs, Hacohen worked with Ryan Corcoran at MGH. Analysis of a clinical trial in which patients with BRAFV600E CRC (mainly MMRp) were treated with a BRAFi, MEKi, and anti-PD-1 showed that the combination improved the objective response rate of both MSS and MSI patients. scRNAseq data further revealed that greater MAPK inhibition correlated with greater induction of immune programs in tumor cells (related to antigen processing and presentation, response to IFNγ and type I IFN, and chemokine activity), increased T cell infiltration, and better outcomes for patients. Matching patient-derived organoids suggested that this effect was driven by tumor-intrinsic innate immune responses, rather than changes in the TME. These results show that improved MAPK suppression can increase immune program induction and enhance antitumor immunity hubs, and Hacohen hypothesizes that even greater induction of these genes could be achieved with ERK inhibition.
Individualized mRNA Neoantigen Vaccines for Pancreatic Cancer - Benjamin D. Greenbaum, PhD – Memorial Sloan Kettering Cancer Center; Vinod Balachandran, MD – Memorial Sloan Kettering Cancer Center
In a tag-team presentation, Benjamin Greenbaum and Vinod Balachandran focused their attention on neoantigens in pancreatic ductal adenocarcinoma (PDAC), a deadly, typically “cold” tumor with poor responses to immune checkpoint blockade and fewer mutations than ICB-responding melanoma or NSCLC. Greenbaum began by summarizing their work analyzing a small subset of PDAC patients who demonstrated unusually long survival (>4-5 years; long-term survivors [LTS]) and for whom there was evidence that immune recognition may have played an important role. Applying rules the group has continued to develop to more critically assess potential neoantigens based on “quality” characteristics (potential for TCR binding, relationship to self, inclusion of frame-shifted neoantigens) of the mutations in these LTS patients led to the conclusions that such patients possessed higher quality neoantigens than typical patients, and interestingly, that relapsed tumors in these patients showed evidence of immune editing of these high-quality neoantigens. The importance of neoantigen-targeted immunity in the LTS population led to an interest in developing personalized neoantigen vaccines more broadly for PDAC patients to boost immunity. Balachandran then described the clinical study that was initiated in collaboration with Genentech and BioNTech in the PDAC adjuvant setting. Neoantigens identified in the primary tumor resection sample were prepared in a liposomal RNA format, with 20 MHC-I and MHC-II neoantigens per patient. After treatment with a single dose of anti-PD-1, patients began a course of 8 intravenous doses of the vaccine product over an 8-week period. Patients then underwent standard adjuvant chemotherapy, and were given a booster dose of the vaccine at week 46. Personal vaccine manufacturing was found to be feasible (delivered 9 weeks after tumor resection) and safe (only one grade 3 adverse event among the 16 patients who received vaccine). Longitudinal TCR repertoire sequencing demonstrated that strong neoantigen-induced T cell responses could be detected, some increasing thousands of fold, and that most were not present prior to immunization in the tumor, suggesting formation of de novo responses. Based on positive results by TCR sequencing results and corresponding ELISPOT results, a “responder” assignment was made for 8 of the 16 patients. Importantly, responders showed high levels of vaccine-induced T cells in the peripheral blood, which persisted for up to 2 years in some cases, and showed a remarkable recurrence-free survival (RFS) advantage compared to non-responders. The absence of other correlates with RFS (response to anti-PD-L1, lymph node or resection margin status, or baseline intratumoral T cell density) suggested a true correlation between vaccine response and RFS advantage.
Library-based Approaches to Detect and Reprogram Immunity - Michael Birnbaum, PhD – Massachusetts Institute of Technology
In the first half of his talk, Michael Birnbaum introduced his team’s lentivirus-based approach for antigen identification. Deconvoluting how pMHCs and TCRs interact will aid in understanding their biology and creating therapies, but high pMHC antigen repertoires along with high TCR repertoires and their very specific and sensitive interactions limit high-throughput studies of such interactions. One of the major limitations is the lack of tools to generate high-throughput interaction data. To solve this issue, the group opted to use lentiviruses for antigen identification and manipulation. Properties such as an understanding of the specific molecular interactions that are required for viral entry into cells, the ability to integrate viral genetic material into host genomes, the ease of generating large lentiviral libraries, and recent advances in viral and molecular engineering have made lentivirus-based antigen discovery feasible. Viruses use VSV-G glycoprotein for binding and fusion, thus facilitating the viral entry. Selected mutations on VSV-G can block the binding, but still retain the fusion capacity. By complementing the binding defect with viral expression of a surface peptide:MHC complex, Birnbaum hypothesized that specific T cells expressing peptide:MHC-specific TCRS could be identified. Using 1G4 TCR Jurkat cell lines, the group provided the proof of concept that off-target pMHC lentivirus did not infect the Jurkat cells, but on-target pMHC lentivirus showed robust infection at low affinities. The group developed an approach called RAPTR (receptor–antigen pairing by targeted retroviruses), which allows creation of peptide/MHCs libraries, each with a barcode that codes peptide/MHC on the viral surface. Following the infection into polyclonal T cells, infected T cells retain the genetic information from the virus, which allows sorting and sequencing to identify the TCR for each pMHC complex. The group validated the concept using NLV peptide and polyclonal T cells. Birnbaum also pointed out that VSV-G viruses can also be used for more general cell entry and genome modification studies in various immune cells. In the second half of his talk, Birnbaum introduced an approach for testing different costimulatory domain combinations in CAR T cells and their functional evaluation (“Carpooling”). The approach encompasses transducing T cells with a CAR library with different domain combinations of potential signaling domains, exposing them to antigen-bearing target cells, and sorting the cells for phenotypes such as proliferation, killing, or change in cytokine production. NGS then allowed the identification of the components of novel candidate CAR designs. Screening costimulatory domains and ITAM domains at membrane proximal positions 1, 2, and 3 showed spatial enrichment patterns for these domains. The group then selected two different CARs (Var1 and Var3) with unconventional domain combinations and showed that the CAR variants can show improved specific lysis, retention of a central memory state, altered cytokine secretion, and persistence in vitro, suggesting that there is a rich amount of phenotypic diversity, which is possible to mine in T cells beyond the usual domain combinations. Interestingly, in vitro functional improvement in Var1 did not translate in vivo for liquid tumors, but did improve tumor control in a solid tumor model, further suggesting that variations in domain combinations can provide solutions for different contextual needs. The platform thus provides rapid identification of CAR signaling combinations at a lower cost, and an opportunity to comprehensively probe how CAR properties, transcriptional programs, and functions interrelate.
Use of High Dimensional Transcriptomic, Proteomic and Spatial Profiling in Mouse Tumor Models of Cancer Immunotherapy and the Applicability of These Studies to Understanding Naturally Occurring and Therapeutically Induced Human Responses to Cancer - Robert Schreiber, PhD, FAIO - Washington University School of Medicine
Mouse models offer numerous strengths in studying and understanding tumor biology and cancer immunotherapies. More than twenty years ago, Robert Schreiber proposed the concept of cancer immunoediting. Studying methylcholanthrene (MCA)-induced sarcomas in immunocompetent or immune-deficient mice, Schreiber found that immunity not only controlled how fast and if tumors grew in these mice, but also shaped the immunogenicity of the cancer cells. Using mouse models and high-dimensional transcriptomic and proteomic analyses over the past years, Schreiber has observed that cancer cells expressing mutations encoding expressed immunogenic neoantigens are rejected early during cancer immunoediting, that some immune-edited tumors with reduced immunogenicity can be controlled by immunotherapy, that mutant MHC class I neoantigens remaining after immunoediting are targets of tumor-specific CTLs following immune checkpoint blockade (ICB), that maximum cancer immunotherapy effectiveness requires T cell responses against both MHC class I and II antigens, and that neoantigen cancer vaccines can be effective as monotherapies and can synergize with ICB in mice. On a detailed mechanistic basis, Schreiber identified distinct lymphoid and myeloid cell subpopulations in progressively growing and therapeutically rejecting T3 MCA sacromas (containing dominant MHC class I and II neoantigens), learned that the subpopulations express distinct transcriptional profiles, functional markers, and sensitivities to different ICBs, and that ICB induce IFNγ from T cells that drives remodeling of both the lymphoid and myeloid compartments of the tumor immune environment. Recent spatial profiling with CODEX revealed that ICB induced the infiltration of the T3 MCA sarcomas with activated CD4+ and CD8+ T cells, which form “islands” with high cytolytic activity in the tumor bed. Having learned so much by using mouse models, Schreiber also knows about the weaknesses of these models, including short time for development of the mouse tumors and TME, a less complex MHC system than in humans, differences in the expression of some mouse versus human checkpoints, and the incompatibility of human ICB drugs and immunocompetent mice.
Cell therapies
A phase I study of personalized adoptive TCR T cell therapy in patients with solid tumors: safety, efficacy, and T cell trafficking to tumor in non-virally gene edited T cells - Stefanie Mandl, PhD - PACT Pharma
Stefenie Mandl reported results from a first-in-human phase I trial of personalized adoptive T cell (ATC) therapy in patients with solid tumors. Personalized ATC therapy starts with the screening, isolation, cloning, and validation of multiple TCRs that recognize patient-specific mutational neoantigens. This is followed by patient enrollment, leukapheresis, and manufacturing of a patient-specific autologous T cell product with a defined composition of up to 3 TCRs. The study design included NeoTCR discovery and selection while patients continued their standard-of-care therapy, including checkpoint inhibitors. Once patients were enrolled on the trial and GMP manufacturing of the personalized ACT had begun, bridging therapy was allowed at the treating physician's discretion. Conditioning chemotherapy prepared the patients for NeoTCR cell infusion on day 0 of the regimen, and tumors were assessed with CT scans at day 28, day 56, and every 2 months for the first year and every three months for the second year. Patients with eight different cancer types were included in the trial, and the study was conducted in two phases: an initial 3+3 dose escalation phase and an expansion phase. Half of the patients in the expansion phase received additional IL-2 for up to 14 doses following NeoTCR infusion. For the eighty-eight patients who were screened on study, more than 50,000 neoantigens presented by 64 different HLA molecules were predicted. For each patient, up to 352 neoantigen-HLA complexes were prioritized and expressed in cell lines, producing around 12,000 patient-specific neoantigen-expressing cell lines. These cell lines were then used to identify TCRs specific for the target neoantigens from patient blood, identifying over 900 unique TCRs. For a breast cancer patient, 102 NeoTCR T cells were identified expressing 15 unique TCRs, which recognized 6 different neoantigen–HLA targets. The 3 best TCRs were selected based on T cell activation status, TCR functionality, neoepitope truncality and expression, and HLA diversity. Non-viral precision genome engineering with CRISPR/Cas9 was used to simultaneously knock out the endogenous TCR and knock in the NeoTCR in autologous T cells, with 40% efficiency of knock-in and 90% efficiency of knock-out in the final cell product. Mandl and colleagues at PACT Pharma were able to expand cells, with yields of up to 10 billion cells for a single NeoTCR product. Out of 88 patients screened for TCRs, 28 got leukapheresis done, and 16 received at least one dose. The regimen was very well tolerated, and the majority of side effects were associated with conditioning chemotherapy. Sequencing data from 8 patients with pre- and post-infusion tumor biopsies showed that NeoTCRs were the top fraction of TCRs in the post-infusion tumors. In one patient with melanoma, the clonal frequency of NeoTCRs at the tumor site went from 0% to 6.4, 1.4, and 1.1% after infusion. Five patients across different tumor types and dose levels achieved stable disease, suggesting antitumor efficacy of the personalized ATC therapy.
Lifileucel TIL cell monotherapy in patients with advanced melanoma after progression on immune checkpoint inhibitors (ICI) and targeted therapy: Pooled analysis of consecutive cohorts (C-144-01 study) - Amod A. Sarnaik, MD, FACS – H. Lee Moffitt Cancer Center & Research
Patients with advanced (unresectable or metastatic) melanoma that has progressed after immune checkpoint inhibition (ICI) or targeted therapy have limited treatment options. Amod Sarnaik reported the results of a single-arm multicenter study lifileucel, an investigational adoptive cell therapy using cryopreserved autologous TIL, representing the largest TIL therapy study in advanced melanoma in the post-ICI setting to date. Lifileucel was produced from harvested tumor specimens using a streamlined 22-day process in 94.7% of the patients. Median time from resection to lifileucel infusion was 33 days. All 153 patients (cohort 2 and 4) received a nonmyeloablative lymphodepletion (NMA-LD) regimen, a single lifileucel infusion, and up to 6 doses of high-dose IL-2. The primary endpoint of the study was investigator-assessed objective response rate per RECIST 1.1 criteria. Across both cohorts, patients had high disease burden, were heavily pretreated with a median of 3 prior lines of therapy, and the majority had primary resistance to prior anti-PD-1 therapy. Treatment-related adverse events were transient, manageable, and consistent with known safety profiles of NMA-LD and IL-2, and incidences decreased rapidly within 2 weeks after lifileucel infusion. The objective response rate was 31.4% , including 9 complete responses (CR) and 39 partial responses (PR). Seventy-one patients had stable disease. Response to lifileucel was observed across all subgroups analyzed, including BRAF mutational status and PD-L1 tumor proportion score. Sum of target lesion diameter above the median and elevated LDH each independently correlated with lower response, suggesting that earlier use of lifileucel in disease progression may provide greater benefit for patients. The median time to response was 1.5 months after infusion. In 7 patients, the initial PR improved to CR, and 10 patients with initial SD improved to PR, suggesting that the responses deepened over time. 41.7% of responses were maintained for at least 24 months, and the median overall survival was 13.9 months.
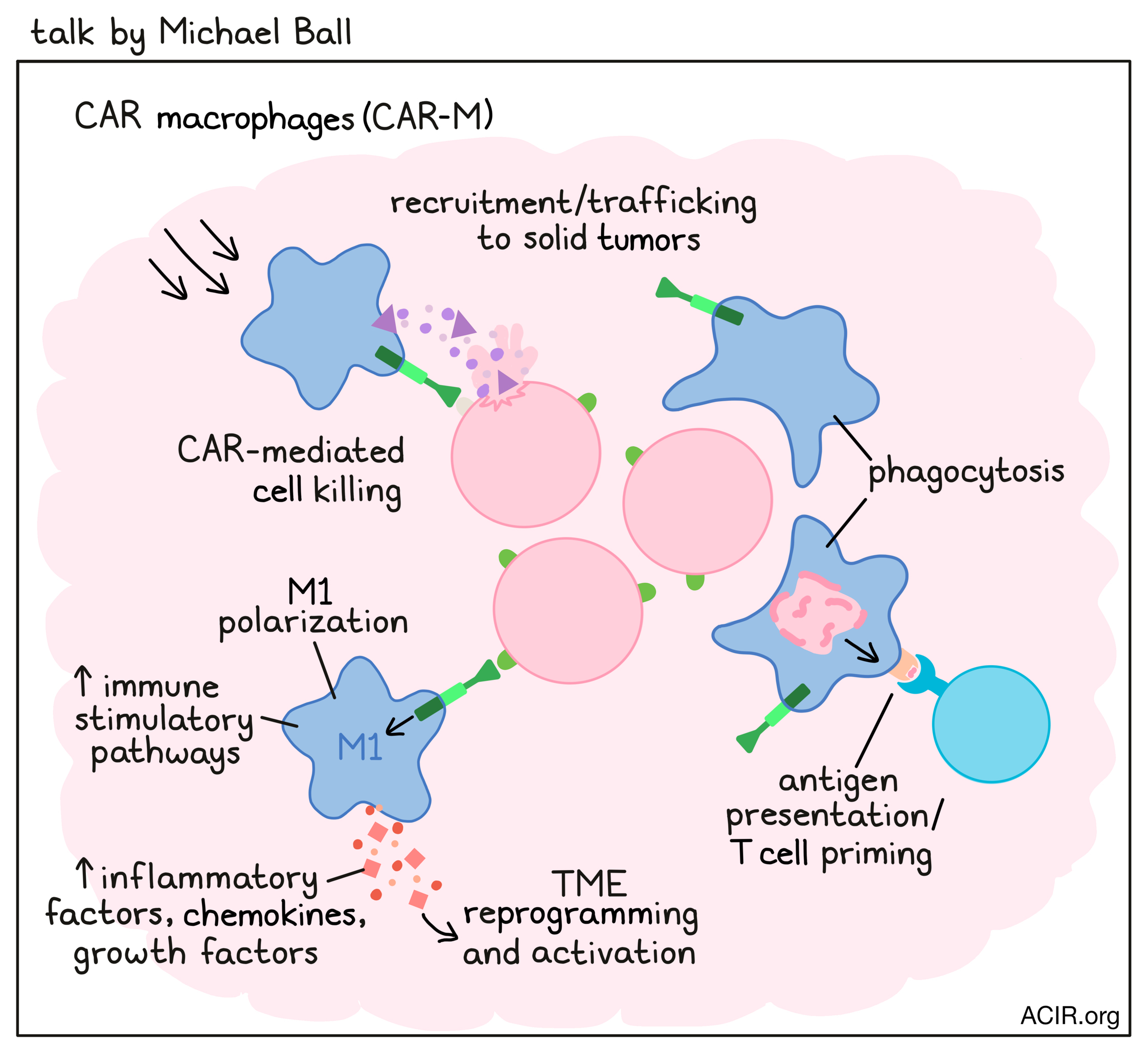
Characterization of CT-0508, an anti-HER2 chimeric antigen receptor macrophage (CAR-M), manufactured from patients enrolled in the phase 1, first in human, clinical trial of CT-508 - Michael Ball, PhD - Carisma Therapeutics
CAR T cell therapy has shown limited efficacy in solid tumors due to poor trafficking to and penetration of the solid tumor, an immunosuppressive and T cell-exhausting TME, and target antigen heterogeneity. Michael Ball presented that CAR macrophages (CAR-M), developed by Carisma Therapeutics, may overcome these challenges. Macrophages are heavily recruited and actively trafficked to solid tumors, where they can, in addition to their CAR function, eliminate tumor cells by phagocytosis. M1-polarized CAR-M can reprogram and activate the TME and, as antigen-presenting cells, prime tumor-reactive T cells. An anti-HER2 CAR-M (CT-0508) product – manufactured from autologous mobilized peripheral blood monocytes within one week, and with a vein-to-vein time of three weeks – is currently being studied in a first-in-human phase 1 clinical trial. CT-0508 could be manufactured for all enrolled patients in group 1 of the trial, with high viability, purity, and CAR expression, similar to products manufactured from healthy donors. Single-cell transcriptomic analysis and surface phenotyping confirmed that each CT-0508 product consisted of M1-polarized macrophages that were capable of specifically killing and phagocytosing HER2+ tumor cells in in vitro assays. Furthermore, antigen engagement potentiated the CT-0508 M1 gene signature and surface phenotype and led to the activation of immune-stimulatory pathways and the production of proinflammatory factors (including TNFα, IL-1β, and IL-18), chemokines, and growth factors that can remodel an immunosuppressive TME.
Checkpoint blockade
Augmentation of CD8 T cell memory via CDK4/6 inhibition can be sequentially combined with PD-1 blockade to avoid toxicities - Stephanie Dougan, PhD – Dana-Farber Cancer Institute
CDK4/6 inhibitors are approved for use in breast cancer and can prolong the survival of patients. However, these cell cycle inhibitors seem to work more like an immunotherapy, with their effect only becoming clear after more than two years of treatment, at the tail of the survival curve. Initial clinical trials investigating CDK4/6i in combination with PD-1 or PD-L1 blockade had to be terminated because of severe toxicity in about half of the treated patients. Stephanie Dougan set out to identify the exact mechanism of action of CDK4/6i and to develop safe combination therapies. Transferred CD8+ T cells that were exposed to CDK4/6i (palbociclib) during priming showed long-term persistence in mice and contributed to immunological memory, effects that were not observed with other cell cycle inhibitors. RNAseq revealed a shift toward memory precursors in CD8+ T cells 48 hrs after activation in the presence of palbociclib in mice and humans, with strong upregulation of MXD4, a negative MYC regulator, and downregulation of MYC targets and cell cycle genes. Dougan and team showed that MXD4 is required for palbociclib-augmented persistence of transferred CD8+ T cells in vivo. The researchers then used scRNAseq to study recently activated human CD8+ T cells and memory precursors, and found that patients who newly started on CDK4/6i as part of their cancer care had higher memory:effector precursor ratios compared to before treatment or to healthy individuals. The finding that CDK4/6 inhibition at the time of priming skewed CD8+ T cells toward a memory fate could be used in manufacturing of adoptive cell therapies, a short course neoadjuvant treatment for resectable tumors, or in combination with vaccines. To study whether there is also a way to safely combine CDK4/6i with PD-1 blockade, Dougan and team went back to the initial clinical trials and performed single-cell profiling of blood samples obtained from patients prior to and on treatment with combination ribociclib and PD-1 blockade. Comparing the results with public datasets as controls, the researchers observed the known effect of PD-1/PD-L1 blockade with new TCR clonotypes coming into the antitumor T cell pool (clonal replacement) and increased proliferation of pre-existing clonotypes. They also found that ribociclib skewed newly emerging clonotypes to a memory cell fate and that the TCRs of these cells from blood also matched TCRs in the tumor. Dougan hypothesized that early exposure to CDK4/6 inhibition can generate a tumor specific memory CD8+ T cell pool that can be later reactivated with checkpoint blockade, and was able to prove this hypothesis in a mouse model of breast cancer.
Dose escalation of next generation Anti-CTLA-4 antibody ONC-392 in combination with fixed dose of pembrolizumab in patients with advanced solid tumors and dose expansion in patients with IO-resistant melanoma - Siwen Hu-Lieskovan, MD, PhD - Huntsman Cancer Institute, University of Utah
While ipilimumab has some clear benefits in a subset of patients, in particular when combined with PD-1/PD-L1 blockade, it is associated with significant toxicities, limiting dose levels and treatment duration. Siwen Hu-Lieskovan discussed ONC-392, a new humanized, pH sensitive, anti-CTLA-4 antibody with a better therapeutic index than ipilimumab. The pH insensitive CTLA-4 inhibitors, such as ipilimumab, lead to CTLA-4 degradation in lysosomes and increase the risk of autoimmune disease. In contrast, ONC-392 avoids lysosomal degradation of CTLA-4 and allows normal recycling of CTLA-4 and the antibody to the cell surface in Tregs, increasing antibody-dependent cell cytotoxicity and reducing immune-related adverse events. The PRESERVE-001 clinical trial assessed the maximum tolerated dose and the recommended phase 2 dose (RP2D) of ONC-392, combined with a fixed dose of pembrolizumab in several solid tumor types. Most patients were heavily pretreated, and many had progressed on previous immune checkpoint blockade (ICB). Two doses were assessed with the lowest dose (3 mg/kg), causing more treatment-related adverse events (TRAE). The recommended phase 2 combination dose was determined to be 6 mg/kg. Out of ten evaluable patients in the dose escalation study, three patients had a partial response (PR), five had stable disease (SD), and two had progressive disease (PD). For the ongoing expansion cohort of patients with advanced melanoma who progressed on previous ICB, preliminary analyses showed that five out of six patients had PR and one patient had SD, with some having long-lasting responses. The combination treatment was generally safe and well tolerated, and the maximum tolerated dose was not reached. Grade 3 TRAEs were observed in five patients; two were infusion reactions and three patients had immune-mediated colitis. This rate of severe immune-related adverse events is considered lower than reported for similar combination strategies.
TIGIT and PD1 are Mechanistically Convergent Checkpoints that Regulate T Cell Differentiation - Ira Mellman, PhD - Genentech
Ira Mellman reviewed data suggesting the TIGIT pathway cooperates with the PD-1 pathway in regulating T cell responses. PD-1/PD-L1 blockade may work at either of two sites; expansion of the T cell compartment or reversal/prevention of T cell exhaustion. Unlike most tumor cells, myeloid cells expressing PD-L1 also express costimulatory ligands. Biochemical reconstitution and in vitro data indicate that PD-1 preferentially regulates the CD28/PI3K pathway, and efficacy of anti-PD-L1 in CT26 tumors requires CD28 signaling. Myeloid cells express most of the PD-L1 expressed in the TME, with macrophages harboring dramatically more (10x) PD-L1 than DCs. PD-L1+ DCs in bladder cancer predict patients with pre-existing immunity, and it is less effective to knockout PD-L1 in macrophages than global or DC-specific knockouts to control murine tumor growth. Therefore, PD-L1 expression on DCs may be key, and PD-1/PD-L1 blockade may act to enhance T cell activation, differentiation, or expansion, rather than reversing exhaustion. Indeed, ATACseq research has shown that exhausted T cells adopt an altered epigenetic state that is largely irreversible. Moving on to the role of TIGIT specifically, and its potential as a therapeutic target for a combination with PD-1/PD-L1 blockade, Mellman noted that TIGIT is expressed early after activation, along with PD-1 on stem cell memory T cells (Tscm), which also express CD226 and CD28. Similar to the CTLA-4, CD28, and CD80/86 story, TIGIT (which contains an inhibitory intracellular ITIM motif) competes with CD226 (which contains an activating intracellular ITSM motif) for PVR (CD155) binding. CD226 is coordinately regulated by TIGIT and PD-1, as TIGIT suppresses CD226 phosphorylation independent of its ITIM domain, and PD-1 blockade enhances CD226 phosphorylation. TIGIT only acts to compete with CD226 for ligand binding, while PD-1 mediates the dephosphorylation of both CD28 and CD226. Therefore, optimal activation of costimulation requires the coordinate inhibition of TIGIT. PD-1A, a phase II trial of the anti-TIGIT antibody tiragolumab combined with atezolizumab (anti-PD-L1), showed promising activity in patients with non-small cell lung cancer. However, the primary progression-free survival endpoint was not achieved in the phase III trial (despite achieving a numerical benefit), but overall survival data are still to be evaluated. Mouse studies have revealed that an intact Fc domain on anti-TIGIT antibodies is required for maximum tumor regression. Therefore, myeloid cells might bind the Fc domain, and through FcγR signaling or structural rearrangements at the synapse, create a more proinflammatory environment. TIGIT and CD226 are also expressed on both Treg and NK cells, and so TIGIT-blocking antibodies may also play a role in these cells. Biomarker data from the clinical trial showed that high infiltration of TAMs and Tregs, along with effector T cells, correlated with best response to the combination therapy. TIGIT and PD-L1 combination blockade may facilitate expansion of antitumor CD8+ T cells, possibly by changing the quality of the CD8+ TIL population – increasing CD226 and decreasing Tox (a key exhaustion transcription factor). This decrease in Tox expression requires the intact Fc domain on the anti-TIGIT antibody. Based on these data, Mellman’s hypothesis is that combined PD-L1/PD-1 and TIGIT blockade redirects the differentiation of activated T cells to an effector or memory phenotype, rather than reversing exhaustion.
Temporal Single-cell Profiling Identifies B-cell Specific Checkpoint Molecules that Regulate Anti-tumor Immunity - Vijay Kuchroo, DVM, PhD – Harvard Medical School
To characterize B cells in B16F10 melanoma tumor, Vijay Kuchroo and his group performed single-cell RNAseq on tumor-infiltrating lymphocytes, tumor-draining lymph nodes, and non-draining lymph nodes from murine tumor samples at days 7, 10, and 18. B cells formed 5 different clusters, representing 2 activated-like clusters, 1 cycling-like cluster, and 2 naive-like clusters. To determine which clusters change during the tumor progression, the groups followed the clusters temporally and observed that over time, cluster 3 expanded only in the draining lymph node in response to the tumor. Expanding B cell cluster 3 expressed Tim-1 and similar cells present on both mouse and human melanoma tumors. Tim-1 is a receptor for phosphatidylserine, and is expressed in only about 10% of B cells. Conditional Tim-1 deletion on B cells initially showed no effect, but as mice aged, resulted in multi-organ tissue inflammation and T cell activation. To determine how Tim-1 loss leads to increased T cell activation, the group isolated B cells from draining lymph nodes and performed bulk RNAseq. This showed that Tim-1+ B cells in draining lymph nodes expressed many known immune checkpoint molecules, like TIGIT, TIM-3, LAG3, and PD-1. To understand the role of checkpoint molecules in B cells, the group generated TIM-3, TIGIT, LAG3, PD-1, and IL-10 knockout (KO) B cells, but didn’t see any difference in tumor growth in these conventional checkpoint molecules or IL-10 KO mice, except very partial growth reduction in TIGIT KO mice. However, conditional TIM-1 deletion in B cells dramatically inhibited tumor growth, with strong induction of CD8+ effector T cell response. Loss of Tim-1 on B cells promoted IFNR1 upregulation, which led to enhanced type-1 interferon responsiveness with increased ability for antigen presentation and T cell help. IFNAR1 blockade in TIM-1 KO tumors abrogated the tumor control, altogether nominating TIM-1 as a potential B cell checkpoint molecule.
Antibody-based therapies
Tumor-Targeted Costimulation via CD28 Bispecific Antibodies: Turning Immunotherapy “Cold” Tumors “Hot” - Dimitri Skokos, PhD – Regeneron Pharmaceuticals, Inc.
Dimitri Skokos discussed the use of various bispecific antibodies for cancer treatment, particularly those that provide both signal 1 to T cells by activation of the TCR/CD3 complex, and signal 2 by engaging CD28 receptors. Various single and combination strategies for bispecific antibodies are currently in clinical trials. The CD20XCD3 bispecific odronextamab (providing signal 1) has been assessed in phase 2 studies, and showed clinical responses, with objective response rates of 53% in aggressive lymphoma and 91% in indolent lymphoma, but improvements can be made. Biopsy samples from a phase 1 trial with odronextamab showed that T cells infiltrated the tumor, that CD28 was preferentially expressed on CD8+ T cells at baseline, and that increases in CD8+ T cells after treatment were mostly driven by the CD28-expressing subpopulation. Combining the CD20XCD3 with the CD22XCD28 bispecific in in vitro cytotoxicity assays showed that CD22XCD28 enhanced cytotoxicity, proliferation, and cytokine secretion, and promoted bystander killing of CD22- diffuse large B cell lymphoma (DLBCL) tumor cells in mixed cultures of both CD22+ and CD22- tumor cells. In a setting where the CD20XCD3 bispecific induced 40% tumor cell killing, adding the CD22XCD28 bispecific improved killing to 80%. In a DLBCL tumor xenograft model, the combination enhanced antitumor efficacy and survival, resulting in an 86% tumor rejection rate, while CD20XCD3 monotherapy had a 0% rejection rate. In mice reconstituted with human immune systems, the combination therapy resulted in the expected depletion of peripheral B cells, expansion of T cells in the periphery, enhanced serum cytokine production, and enhanced survival compared to either monotherapy. Immune profiling showed increases in intratumoral CD8+ T cells, which were largely of an effector memory phenotype. Combination therapy enhanced reprogrammable CD8+ T cells and decreased dysfunctional CD8+ and CD4+ T cells. Finally, the combination was tested in cynomolgus monkeys, where it efficiently depleted peripheral B cells and synergistically activated T cells, with no adverse events reported after repeated dosing. Therefore, CD28 costimulatory bispecific antibodies, delivering critical signal 2, may serve as off-the-shelf T cell response boosters that can be combined with current bispecific therapies to overcome resistance and improve efficacy.
Tumor-targeted Delivery of Immunostimulatory Cytokine/Antibody Fusion Proteins - Jamie Spangler, PhD – Johns Hopkins
Cytokines induce receptor dimerization to regulate immune activity, allowing the intracellular protein to come together and initiate the cascade of events that culminates in nuclear transcription. Jamie Spangler focused on interleukin 2, which is distinct from many other cytokines, as it can signal through dimeric (β and γ) or trimeric (α, β, and γ) receptor complexes. IL-2 has a 100-fold higher affinity for the trimeric (10 pM) than the dimeric (1nM) receptor complexes. Different cells express IL-2 receptors at variable levels, with the dimeric receptors expressed on effector cells, whereas the trimeric receptors are expressed on regulatory T cells (Tregs). High doses of IL-2 are used in cancer immunotherapy to activate effector cells with lower IL-2 affinity, leading to severe toxicity, whereas low doses of IL-2 are used for Treg activation to treat autoimmune diseases. For mice, two specific antibodies have been discovered to selectively bias IL-2 activity, S4B6-IL-2 towards activation of effector cells and JES6-1-IL2 towards Tregs. However, the molecular basis of this bias was previously unknown. The crystal structure of IL-2-bound S4B6 antibody, which biases towards the effector cells, showed that the antibody blocks the IL-2Rα subunit, but not the β or γ subunits. S4B6 mimics IL-2Rα to allosterically enhance IL-2Rβ binding on effector cells. However, in Tregs, S4B6 blocks IL-2Rα, decreasing IL-2Rα binding-mediated enhanced affinity. Due to the limitations of cytokine–antibody complexes, such as toxicity due to complex dissociation, dosing, and increased regulatory hurdles, the group designed an intramolecular human immunocytokine (IC) (602), where the antibody physically binds to IL-2. Although the two antibodies (S4B6 and 602) shared similar mechanisms structurally, 602 showed less bias towards effector cells than S4B6. The group then went on to engineer 602 into the F10 immunocytokine by mutagenesis to make it more competitive with IL-2Rα and gain greater bias towards effector cells. F10 IC was more competitive against IL-2Rα, showed higher affinity for IL-2Rβ, increased bias towards effector cells, and preferentially activated human CD8+ T cells. F10 IC treatment expanded CD8+ T cells and NK cells relative to Tregs in C57BL/6 mice, and reduced tumor growth in the B16F10 and CT26 tumor models. To direct the immunocytokine towards tumors, Spangler’s group fused F10 IC with the collagen binding domain (CBD). IC/CBD fusion bound to human collagen with higher specificity, improved its tumor retention, and inhibited tumor growth without toxicity compared to F10 IC. This demonstrated that cytokine/antibody fusion enables selective delivery of IL-2 in a stable and translationally relevant format, and that collagen binding domains allow targeted delivery of the immunocytokine to the tumor microenvironment.
Tumor microenvironment
Commensal microbiota can impact anti-tumor immunity at the level of myeloid cells - Thomas Gajewski, MD, PhD – University of Chicago
Thomas Gajewski’s presentation began by distinguishing T cell-inflamed and non-inflamed tumors, which differ in CD8+ T cell infiltration, chemokines, type I IFN gene signatures, and more. Under these differential immune pressures, tumors utilize different mechanisms of immune escape, with non-inflamed tumors escaping via immune exclusion, and inflamed tumors escaping via antigen loss or utilization of immune-inhibitory pathways. Compared to non-responders, most patients who respond to anti-PD1 checkpoint blockade have a T cell-inflamed tumor microenvironment signature at baseline. Curious as to what factors might contribute to these differences between patients, Gajewski and colleagues investigated potential sources of inter-patient heterogeneity of the T cell-inflamed tumor microenvironment, with consideration towards tumor cell somatic differences (Wnt/β-catenin activation, PTEN loss, DNA repair machinery), environmental differences (commensal microbiota), and host germline genetic differences in immune regulatory genes (PKCδ variants). Focusing his talk on the environmental factors, Gajewski reviewed the data demonstrating that the composition of the gut microbiome is associated with the efficacy of PD-1 blockade in metastatic melanoma patients and that anti-PDL-1 is effective in germ-free mice when reconstituted with human responder microbiota, but not non-responder microbiota. To better study this mechanism and avoid the “shock” of acute bacterial transfer into adult germ-free mice, Gajewski and colleagues developed reverse-translational mouse models (“avatar” mice), in which germ-free mice that had been reconstituted with microbiomes from responder and non-responder patients were bred, yielding progeny that stably maintained the parental gut microbiomes. Fecal microbiota transplant (FMT) back and forth between mice with responder and non-responder microbiomes revealed that non-responder microbiome mice could benefit from transfer of gut microbes from responder microbiome mice, and similarly, responder microbiome mice could be impaired by the transfer of gut microbes from non-responder microbiome mice. These results suggested the presence of both “good” and “bad” microbes that could influence outcomes. Delving into the underlying mechanisms, the researchers found that in the presence of a favorable microbiome, the macrophage population in the tumor microenvironment was enriched for M1-like genes, versus the M2-like genes observed in tumors in mice with an unfavorable microbiome. In the granulocyte compartment, responder microbiome mice were enriched for neutrophil-like genes, while non-responder microbiome mice were enriched for MDSC-like gene expression. Further, an M1:M2 ratio signature derived from this mouse data showed that patients who were non-responders to PD-1 blockade had a low M1:M2 ratio, suggesting that the patterns identified in mice may be consistent in patients. Efforts are ongoing to identify the characteristics of favorable bacteria in responder mice using tumor control in non-responder avatar mice as a readout.
Mouse Models of Human Immune Tumor Microenvironments - Anthony Rongvaux, PhD – Fred Hutchinson Cancer Research Center
Anthony Rongvaux generated two different strains of “humanized mice” by intrahepatic injection of human CD34+ hematopoietic stem and progenitor cells (HSPCs) into irradiated newborn MISTRG or ISTRG mice. MISTRG mice are immunodeficient (RAG2-/- and IL2Rγ-/-) and have human cytokine genes knocked into the respective murine endogenous locus (M-CSFh/h, IL-3/GM-CSFh/h, SIRPαh/m, TPOh/h). ISTRG mice are similar, but lack human M-CSF, which is important for myeloid cell differentiation. After 3 to 4 months, more than half of the white blood cells were of human origin in these mice. MSTRG mice support the development of all major immune cell lineages, while ISTRG mice have fewer monocytes in the blood and almost no macrophages in tissues. An implanted human BRAFV600E melanoma cell line (Me275) showed drastic differences in disease progression in MISTRG and ISTRG mice, recapitulating the tumor-supporting function of macrophages. While Me275 melanoma formed metastases in liver, lung, and spleen in MISTRG mice, ISTRG mice had very few metastases. By transcriptional analysis, tumor-infiltrating macrophages of MISTRG mice were very similar to macrophages from human tumors, and different from murine cells described as M2 macrophages or MDSCs. T cells in Me275-bearing MISTRG mice were localized at the tumor margin and co-localized with a large number of macrophages. Interestingly, T cell exclusion was not dependent on macrophages, but a tumor-intrinsic property. Rongvaux and colleagues derived two gene signatures for hot versus cold tumors and applied these to a published dataset of over 40 melanoma cell lines. Four of these cell lines predicted to be hot or cold were inoculated in their humanized mouse models and proved that the transcriptional signatures of cold and hot tumor cell lines were predictive of T cell exclusion in the humanized mice. Thus, Rongvaux effectively demonstrated that MISTRG mice recapitulate human immune tumor microenvironments and can be used to study human tumor biology and modifications thereof.
Utilizing Tumor Microenvironment Antigens for Immune Modulatory Vaccines - Mads Hald Andersen, PhD – Herlev and Gentofte Hospital
More than ten years ago, Mads Hald Andersen and his team first described spontaneous IDO-specific T cell responses in patients with cancer. This was the first example of a group of effector cells, called anti-regulatory T cells, that were pro-inflammatory and could modify a suppressive tumor microenvironment via recognition and attack of suppressive immune regulatory cells (e.g., myeloid cells, Tregs, DCs, and CAFs) expressing and presenting the cognate antigen (e.g., IDO, PD-L1, Arginase, and Foxp3). A phase I clinical trial of an HLA-A*02-restricted IDO peptide vaccine in heavily pre-treated patients with late-stage non-small cell lung cancer showed a signal of extended survival compared to non-vaccinated (HLA-A*02-negative) patients. The long-term survivors after IDO peptide vaccination are now disease-free for more than 7 years. Therapeutic vaccination with IDO peptide and an anti-PD-1 antibody showed clear synergy and eliminated IDO-expressing immunoregulatory myeloid cells in a mouse CT26 tumor model. After more encouraging results from a phase I trial of a PD-L1 peptide vaccine in incurable multiple myeloma patients that led to immune responses to PD-L1 in all patients, Andersen launched a phase I/II trial of biweekly IDO and PD-L1 peptide vaccination combined with anti-PD-1 therapy as a first-line treatment of melanoma. Combination therapy resulted in an increase in complete and partial responses compared with a strongly matched historical control cohort, and side effects were comparable to nivolumab monotherapy. CD4+ (predominant) and CD8+ T cell responses were observed to both antigens, and TCR sequencing of paired biopsies demonstrated an increase in target-specific T cells at the tumor site in 4 of 5 patients, irrespective of response. Further, target-specific T cells could recognize autologous tumor cells or myeloid cells cultured with tumor cell-conditioned medium to create a TAM phenotype with increased expression of PD-L1 and IDO in vitro. A phase 3 trial is currently ongoing. Overall, this novel approach opens up multiple new target opportunities. Andersen also touched briefly on a TFGβ and Arg-1 vaccine stemming from his lab.
In situ vaccination and cancer vaccines
Personalized DNA neoantigen vaccine (GNOS-PV02) in combination with plasmid IL-12 and pembrolizumab as second-line (2L) treatment for advanced hepatocellular carcinoma (HCC) - Edward J. Gane, MD - The University of Aukland
Edward Gane reported on the safety, efficacy, and immune response of the personalized DNA neoantigen vaccine, GNOS-PV02, which was investigated in a phase 1b/2a multicenter study (GT-30) in patients with advanced hepatocellular carcinoma (HCC), who progressed during or were intolerant to first-line tyrosine kinase inhibitor (TKI) treatment (though atezolizumab plus bevacizumab has recently replaced TKIs as the first-line treatment of HCC). The response rate for the current second-line treatment with anti-PD-1 immunotherapy, however, is poor at only 16% (KEYNOTE394). To address this need, Gane and colleagues developed a personalized DNA neoantigen vaccine. The GNOS-PV02 vaccine has three components: an optimized large DNA plasmid encoding up to 40 neoantigens (which is sometimes all the neoantigens identified for a patient), a plasmid encoding IL-12 cytokine adjuvant, and a CELLECTRA in vivo electroporation device for intradermal delivery. Needle-to-needle production time was approximately 6-8 weeks. The vaccine was delivered together with anti-PD-1 therapy every 3 weeks for several cycles, and then the vaccine alone was continued. The GNOS-PV02 vaccine plus pembrolizumab combination was well tolerated, and the vaccine-related AEs were all mild and resolved by themselves with time. Out of 23 patients evaluable for efficacy, 2 had complete responses (CRs), 5 had partial responses (PRs), and 6 had stable disease (SD) with an overall response rate of 30.4%. One of the CR patients, a 73-year-old white male with HCC harboring a beta-catenin mutation (CTNNB1 S45F) associated with resistance to immune checkpoint blockade, demonstrated complete tumor reduction and rapid disappearance of circulating tumor DNA over a 7-month period, and his cancer remains undetectable more than 1 year later. ELISPOT and flow cytometry demonstrated that the GNOS-PV02 vaccine induced new and expanded neoantigen-specific CD4+ and CD8+ T cells in all patients. TCR sequencing showed that new T cell clones could be detected in both the peripheral blood and tumor tissue. Based on these promising results, the current study has been expanded to 36 patients.
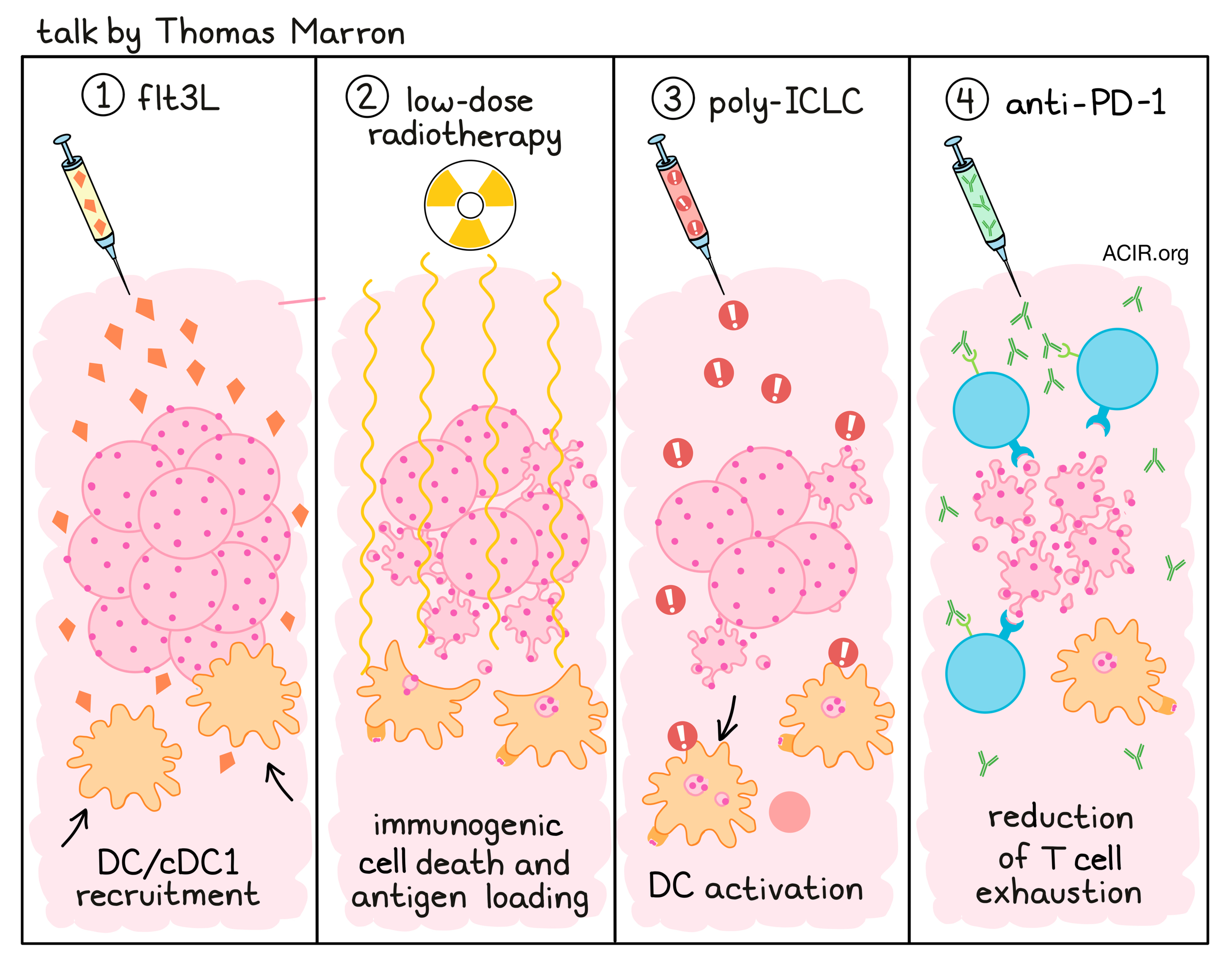
Flt3L-primed in situ vaccination and pembrolizumab induce systemic tumor regressions of bulky tumors in patients with lymphomas and ER/PR+ breast cancer - Thomas Marron, MD, PhD – Icahn School of Medicine at Mount Sinai
Most patients do not respond to immune checkpoint blockade (ICB), and having immune “hot” tumors is not always enough for therapeutic efficacy. Thomas Marron reviewed the need for dendritic cells, in particular cDC1s, to prime de novo responses for effective ICB. To induce such responses, a vaccination strategy was developed to augment cDC1 using Flt3L, which can be added to classic adjuvants such as poly-ICLC to augment in situ vaccination. Preclinical studies showed that injection with Flt3L recruits DCs to the tumor site. Then low-dose radiotherapy can be used to generate immunogenic cell death and load the DCs with antigen, and finally, treatment with poly-ICLC activates the DCs. This triplet therapy is potent in mice. In a past clinical trial conducted in patients with non-Hodgkin lymphoma (NHL), intratumoral Flt3L and poly-ICLC were combined with low-dose radiotherapy. This resulted in a ten-fold increase in cDC1 with a third of patients showing good responses, including a durable CR. However, the majority of patients did not respond and their cancer recurred. Tumor tissue of one treated non-responding patient showed robust T cell infiltration after therapy, but with significant exhaustion signatures in both CD8+ and CD4+ T cells. Based on this, mouse experiments were conducted that combined the triplet treatment with anti-PD-1, which improved responses. In the currently presented clinical trial, patients with indolent NHL (N=3), metastatic breast cancer (MBC; 4/6 hormone positive), or head and neck squamous cell carcinoma (N=1) who failed at least one first-line therapy were treated with radiation and Flt3L, followed by poly-ICLC and pembrolizumab (anti-PD-1). Treatment was well tolerated, except for adverse events expected for poly-ICLC. The ORR was 30%, with 1 complete response (NHL), 2 partial responses (MBC), 1 stable disease, and 6 progressive disease. The responders included a patient with diffuse large B cell lymphoma (DLBCL) who was without evidence of disease two years after treatment was initiated; the response rate to ICB alone is 4% in this cancer type. Two patients with hormone receptor-positive breast cancer who previously had multiple lines of chemotherapy showed response to treatment, including abscopal responses, even though this cancer type generally does not respond to immunotherapy. Assessing markers for response at the time of completion of the vaccine dosing, before completion of pembrolizumab, there was a difference in chemokine levels, and DC1s increased in responders (but not non-responders). TCR repertoire analysis showed that some pre-existing clones significantly expanded in responders, and priming of de novo clones was also observed.
Oncolytic viruses
Oncolytic Viral Therapy - Beyond the 1st Generation - Igor Puzanov, MD, MSCI, FACP – Roswell Park Comprehensive Cancer Center
The first-generation oncolytic virus T-VEC was approved for treatment of stage IIIB-IV melanoma in 2015. Unfortunately, a phase III clinical trial investigating the combination of locoregional T-VEC with pembrolizumab in patients with melanoma (MASTERKEY-265) yielded no significant benefit in progression-free survival (PFS) or overall survival (OS), and was stopped in 2021. To understand why this trial failed, Igor Puzanov looked back at the baseline patient characteristics and found that the patients enrolled in the trial had disease that was "relatively easy" to treat by monotherapy, and did not necessarily require combination therapy. Further, Puzanov found that patients who were treated at a trial site in the USA had higher PFS, favoring the T-VEC arm more than in patients treated on the trial outside the USA. According to Puzanov, US investigators had prior experience with intratumoral oncolytic virus injection, while other centers were less experienced. Questioning whether anti-PD-1 was the ideal combination partner for T-VEC, Puzanov described the results of a randomized phase II trial of ipilimumab with locoreginal T-VEC as first-line treatment for stage IIIB-IV melanoma. This trial met its primary endpoint, with an objective response rate of 39% in the combination cohort versus 18% in the ipilimumab monotherapy group. Puzanov also discussed how the antitumor efficacy of T-VEC, which is based on a herpes simplex virus 1 backbone, could be enhanced with additional payloads. ONCR-177, also based on HSV-1, is armed with 5 payloads including IL-12, FLT3L, CCL4, anti-CTLA-4, and anti-PD-1, compared to T-VEC, which only carries GM-CSF. In a phase 1 study ONCR-177 led to regression of a resistant squamous cell head and neck tumor. RP-1, which is based on T-VEC but has an optimized backbone, has been efficacious against tumors that were resistant to T-VEC with deep and durable responses in patients with non-melanoma skin cancer and patients with melanoma who had previously failed on anti-CTLA-4/anti-PD1, with abscopal effects observed in uninjected metastases (e.g., bone). Currently, Puzanov is evaluating a Newcastle disease oncolytic virus that incorporates IL-12 (MEDI9253) in a phase I clinical study in combination with durvalumab in patients with advanced CRC, RCC, or melanoma. But he also pointed out a recent setback in the field in which the development of CAVATAK, a coxsackie-based oncolytic virus, was stopped after neutralizing antibodies against the virus were observed following i.v. administration. Self-amplifying viral RNA encapsulated in lipid nanoparticles may serve as a solution to allow for repeated i.v. administration. The best way forward for oncolytic virus-based immunotherapy remains unclear, but improvements to the backbone and payload, standardization and design of injection techniques, as well as patient selection and biomarker development will be essential when aiming to improve responses.
By Ute Burkhardt, Ed Fritsch, Lauren Hitchings, Shishir Pant, and Maartje Wouters



