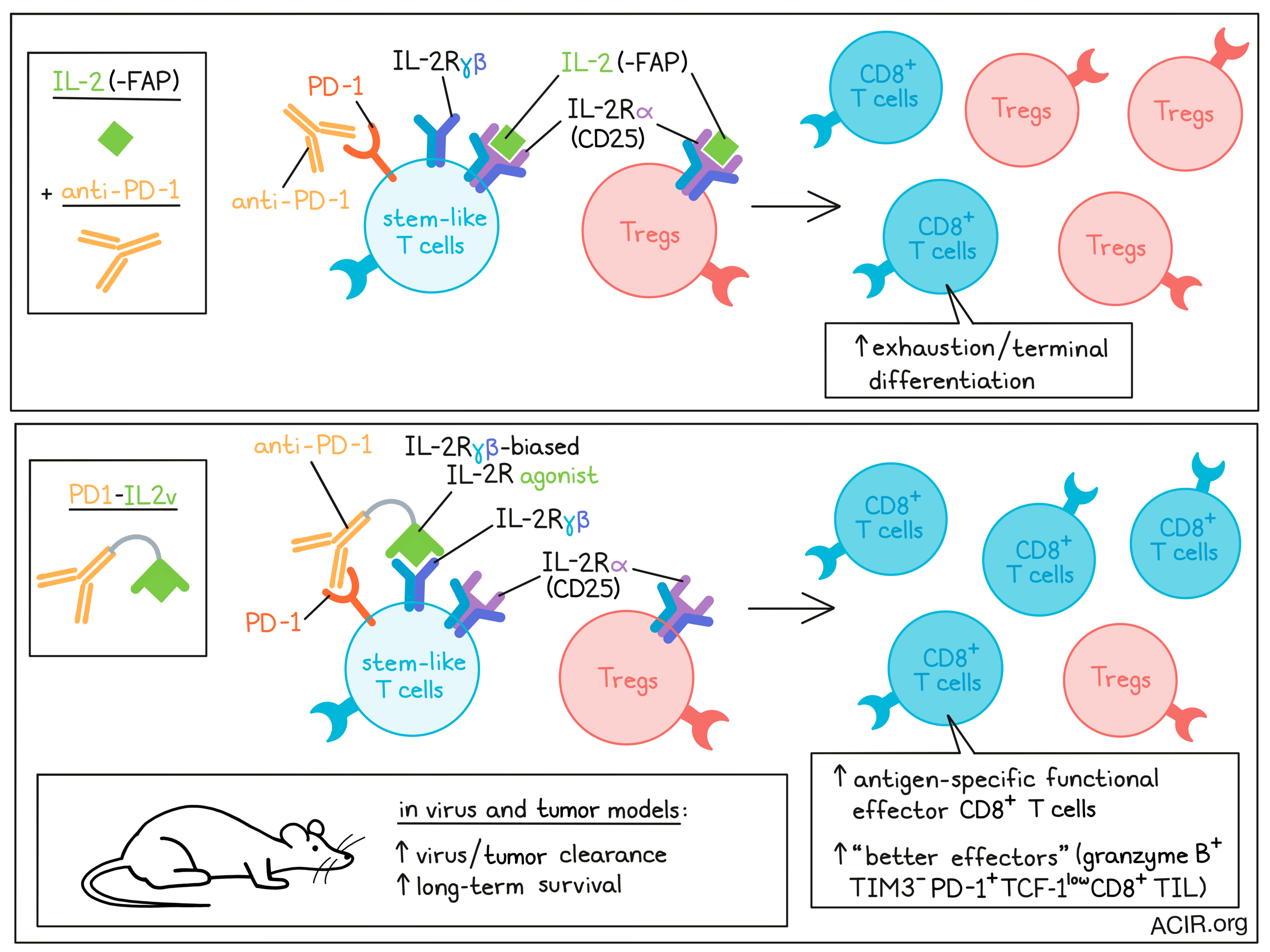
IL-2 can induce the differentiation of stem-like T cells into CD8+ T cell populations with better effector functions in chronic infection. However, systemic treatment with IL-2 induces adverse effects, such as the expansion of Tregs. To overcome this problem, Deak, Nicolini, Hashimoto, and Karagianni et al. engineered an immunocytokine, PD1-IL2v, that induces stronger and more exclusive expansion of better effector cells. This therapy was tested in chronic infection and cancer models, and the results were recently published in Nature.
The researchers designed PD1-IL2v to provide IL-2R agonism preferentially to PD-1+ tumor- or virus-reactive T cells, linking a PD-1-binding antibody to an IL-2 receptor β- and γ-chain-biased agonist, thus simultaneously blocking the PD-1 pathway while agonizing IL-2R on the same cell. PD1-IL2v was found to be 40 times more potent than control fibroblast activation protein-targeted FAP-IL2v in vitro. In vitro cis versus trans assays with membrane-labeled CD4+ T cells showed that the frequency of pSTAT5+ T cells, representing IL-2R signaling, was lower in cells that were pre-blocked with saturating doses of anti-PD-1 prior to mixing with unblocked cells and exposure to PD1-IL2v, even though they were in close proximity to cells that were not blocked. This suggested that PD1-IL2v induced IL-2R agonism in cis on the same cells after binding PD-1.
To assess whether PD1-IL2v would have stimulating effects on Tregs, binding competition and suppression assays with Tregs and conventional CD4+ T (Tconv) cells were performed. PD1-IL2v bound preferentially to the Tconv, which had higher levels of PD-1 than Tregs. The binding of PD1-IL2v to Tconv helped these cells overcome Treg-mediated suppression in a dose-dependent manner.
The researchers then investigated the internalization of PD1-IL2v and control FAP-IL2v using fluorescently labeled versions in an assay with PD-1+CD4+ T cells. PD1-IL2v (which bound both PD-1 and the IL-2R) was internalized with slower kinetics than FAP-IL2v (which bound only IL-2R), resulting in potentially longer IL-2 signaling. Bound PD-1 receptors were co-internalized with IL-2R. The researchers found that PD1-IL2v could also induce GM-CSF and granzyme B secretion in PD-1+CD4+ T cells in a dose-dependent manner, as potently as wild-type IL-2.
The researchers then assessed whether PD1-IL2v could deliver IL-2v to lymphocytic choriomeningitis virus (LCMV)-specific CD8+ T cells in vivo during chronic infection. The combination of anti-PD-L1 and PD1-IL2v was superior to anti-PD-L1 monotherapy in increasing the number of LCMV-specific CD8+ T cells in various tissues. These specific T cells had a polyfunctional signature (producing IFNγ, TNFα, and/or IL-2) and therapy-induced changes in the expression of phenotypic markers. RNAseq of these cells showed that this regimen changed the transcriptional signature, including increased expression of Cd28, various cytokine receptors, chemokines related to T cell migration, adhesion molecules, and molecules related to egress from lymphoid tissues. These are features of functional effector CD8+ T cells, while exhausted T cell markers and transcription factors such as Tox and Pdcd1 were downregulated. This phenotypic change resulted in better viral control. PD1-IL2v monotherapy also induced expansion of LCMV-specific CD8+ T cells, but the combination treatment increased numbers more and resulted in more marked phenotypic changes. Similar highly functional T cells were observed in a LCMV chronic infection model following anti-PD-1 + IL-2 therapy[1].
Adoptive transfer experiments were performed to assess which CD8+ T cell population is targeted by PD1-IL2v. Stem-like (PD-1+CXCR5+TIM3-) and terminally-differentiated (exhausted; PD-1+CXCR5-TIM3+) CD8+ T cells were sorted from chronically LCMV-infected mice and transferred into infection-matched mice, after which mice were treated with PD1-IL2v. In this model, only the stem-like CD8+ T cell subset proliferated. Treatment induced optimal effector differentiation, as shown by the upregulation of CD218a with low to intermediate TIM3 expression.
Moving to cancer models, C57BL/6 mice were first inoculated orthotopically with Panc02-H7-Fluc pancreatic adenocarcinoma cells and treated with PD1-IL2v or controls. PD1-IL2v induced tumor eradication and resulted in long-term survival, while controls did not cure the mice. Induced T cells were then studied in subcutaneously implanted tumors. Treatment with PD-1-IL2v generated and expanded effector memory TIL, and CD8+ TIL induced by therapy were polyfunctional and co-expressed higher levels of granzyme B, IFNγ, and TNFα than TIL from control groups. In the periphery, these effects were not detected, confirming the importance of high PD-1 expression levels for therapy effects. Depletion of CD8+ T cells prevented tumor growth inhibition, confirming the requirement for CD8+ TIL for treatment efficacy. Tumors had an effector memory CD8+ T cell population with much higher levels of PD-1, while IL-2Rβ frequencies and receptor numbers per T cell were similar in tumors and blood.
To assess antitumor effects, mice bearing subcutaneous Panc02-H7-Fluc tumors were then treated with PD1-IL2v, high-dose IL-2, or either monotherapy combined with pembrolizumab (anti-PD-1). Survival and tumor growth reduction were more pronounced in mice treated with PD1-IL2v monotherapy than those treated with the combination of high-dose IL-2 and pembrolizumab. The combination of PD1-IL2v and pembrolizumab did not impair the efficacy, but also did not induce additional benefit.
Treatment with PD1-IL2v increased the frequency of the granzyme B+TIM3- population within the PD-1+TCF-1low/- CD8+ TIL, which the researchers called “better effectors”. scRNAseq of these better effectors showed that these expressed pro-inflammatory and homeostatic proliferation and memory formation cytokine signatures, as well as high levels of Pdcd1, intermediate levels of Lag3, and low levels of Havcr2, Tigit, and Tox, matching a non-exhausted phenotype. Furthermore, these TIL had higher stem-like and migration signature scores. The better effector CD8+ TIL also showed a high overlap in clonotypes with stem-like CD8+ TILs and the highest number of expanded clones, suggesting tumor specificity.
Finally, the researchers assessed the efficacy of PD1-IL2v in mice implanted with the B16F10-OVA syngeneic cell line. Treatment provided longer survival to half of the animals, and 20% of mice were cured. Therapy expanded the population of OVA-specific CD8+ TIL, as well as OVA-specific PD-1+TCF-1+ stem-like T cells in the tumor. Therapy induced expansion of granzyme B+TIM3- and granzyme B+TIM3+ populations within the OVA-specific PD-1+TCF-1low/-CD8+ TIL. Two additional mouse models, MCA-205 sarcoma, which is partially sensitive to PD-1 blockade, and RIP-Tag5, which is unresponsive to PD-1 inhibition, were also assessed for therapy efficacy. In both models, PD1-IL2v resulted in tumor control and survival benefit (albeit with some toxicity due to hypoglycemia in the RIP-Tag model (rat insulin promotor driving SV40 T antigen).
These data suggest that this novel PD-1-linked IL-2 agonist can have beneficial tumor-specific effects. It not only blocks the PD-1/PD-L1 axis, but also agonizes T cells and results in an effector phenotype that may be able to overcome immune inhibition in both chronic infections and cancer.
[1] https://doi.org/10.1038/s41586-022-05257-0
Write-up by Maartje Wouters, image by Lauren Hitchings.
Meet the researcher
This week, first co-author Laura Codarri Deak and lead author Pablo Umaña answered our questions.

What was the most surprising finding of this study for you?
LCD and PU: We designed PD1-IL2v to deliver IL-2R agonism to PD-1+ T cells, a bona fide marker of antigen-specific T cells, and already in early experiments, it showed remarkable efficacy, even in challenging tumor models. However, the discovery and unravelling of its mechanism of action has taken major efforts in the design of experiments aiming to dissect its properties. An unexpected finding via our collaboration with Rafi Ahmed and his team at Emory University, was that pure IL-2Rβγ-biased agonists lacking CD25 binding cannot trigger an alternative differentiation path of the stem-like, PD-1+CD8+ T cells into “better effectors” that are more cytotoxic and more resistant to exhaustion. However, we were able to overcome that, while avoiding the undesirable aspects of CD25 binding, by targeting IL2v in cis to PD-1. In other words, we redirect enhanced IL-2R agonism from being guided by CD25 to instead follow PD-1 expression.
What is the outlook?
LCD and PU: While we were among the first to bring strict IL-2Rβγ-biased agonists into the clinic with CEA-IL2v and FAP-IL2v, we learnt from the clinic that their efficacy was inferior to what one would have expected. Our preclinical findings showed the important role of CD25 binding for the biology of wild-type IL-2, and how this can be applied to design the novel PD-1 cis-targeted IL2v immunocytokine with its unique mode of action. We believe that PD-1 cis-targeted IL-2 can be a major step towards improved immunotherapies, and it is already showing promising signs of clinical activity. In general, this particular approach of delivering cytokines to immune cells is an important advance in the field of immunotherapy, allowing for the preferential modulation of particular subsets of the immune system to reduce or eliminate the disease burden.
What was the coolest thing you’ve learned (about) recently outside of work?
LCD: I love open water swimming and snorkeling, and I always enjoyed traveling far to tropical and exotic places to see new underwater life. I was recently surprised to find similar creatures in the Mediterranean sea, where I spend most of my childhood and teenage summers. This reminds me that sometimes we have what we are looking for right in front of our eyes, but we need to be “ready” and “have an open mind” to be able to see it.



