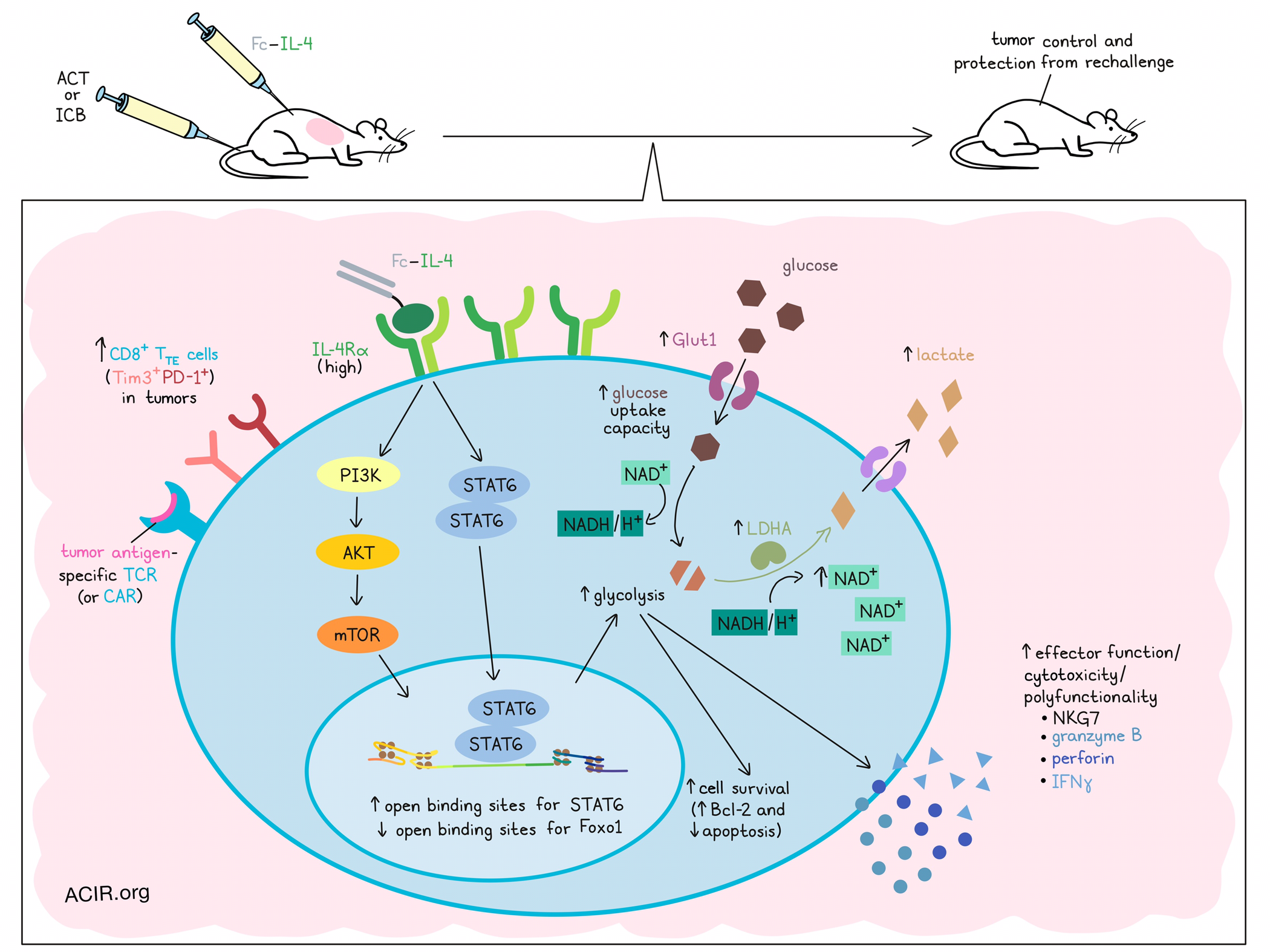
Interleukin 4 (IL-4), a cytokine typically associated with humoral (type 2) immune responses, has recently been shown to promote survival in T and B cells. Studying whether IL-4 could be exploited to enhance cancer immunotherapy, Feng and Bai et al. developed a fusion protein of IL-4 and a mutant IgG2a antibody (Fc–IL-4), and tested its effects across various settings. Their results, recently published in Nature, showed that Fc–IL-4 induced direct metabolic reprogramming in terminally exhausted CD8+ T cells, reinvigorating their functionality and enhancing antitumor immunity, especially in combination with existing immunotherapies.
To begin, the researchers evaluated Fc–IL-4 in the context of mice bearing B16F10 tumors (expressing gp100) and treated with adoptive cell transfer (ACT) of PMEL T cells (targeting gp100). Here, the addition of Fc–IL-4 increased both PMEL and endogenous T cells in tumors, and enhanced their polyfunctionality and production of granzyme B. Among PMEL T cells, the PD-1+Tim3+ (TTE) subset was enriched. These effects were restricted to tumor antigen-specific T cells, as they were not observed for OT-1 cells transferred to non-OVA-expressing tumors.
Investigating changes to immune cells in tumors, the researchers performed scRNAseq and sorted tumor antigen-specific Thy1.1+CD8+ TILs from tumors from ACT- and Fc–IL-4-treated mice. Several of the clusters that emerged resembled TTE cells, with high expression of co-inhibitory molecules, while 1 showed attenuated expression of these markers. After Fc–IL-4 treatment, the more TTE-like clusters increased, while the less exhausted cluster was limited. Expression of Ifng, Gzmb, Prf1, and Nkg7 were also higher in treated samples, suggesting enhanced cytotoxic activity.
Next, the researchers evaluated whether Fc–IL-4 could enhance antitumor immunity, and found that across numerous murine tumor models, the addition of Fc–IL-4 to ACT significantly improved antitumor responses, leading to durable cures and protection from rechallenge. Similar results were observed using a human Fc–IL-4 in xenograft mouse models treated with human CAR T cells, including a model of recurrent leukemia. In some models, Fc–IL-4 showed antitumor activity on its own, suggesting that it supports endogenous antitumor immune responses as well. Lymphodepletion was not required for antitumor efficacy, and no overt toxicities associated with Fc–IL-4 were observed. Fc–IL-4 also enhanced the antitumor efficacy of immune checkpoint blockade (ICB; anti-PD-1 + anti-CTLA-4) in an MC38 tumor model, where it led to clearance of established tumors and protection from rechallenge.
Selective depletion studies revealed that CD8+ T cells were essential to the antitumor efficacy of Fc–IL-4, while progenitor exhausted T cells were dispensable. By transferring different T cell subsets for ACT, the researchers found that only mice treated with PD-1+TIM-3+ CD8+ TTE cells showed enhanced enrichment and effector functions in response to Fc–IL-4, suggesting Fc–IL-4 acts mainly on this subset. In line with this, CD8+ TTE cells expressed the highest levels of IL-4Rɑ among all evaluated subsets, and IL-4Rɑ-KO CD8+ TTE cells failed to respond to Fc–IL-4, suggesting that Fc–IL-4 acts through direct signaling on CD8+ TTE cells. The use of FTY720 to block T cell egress from lymph nodes had little impact on the effects of Fc–IL-4, suggesting that it acts primarily on cells within tumors, rather than on cells recruited from the periphery.
The researchers found that while neutralizing IL-4 in tumor models showed little impact, the addition of Fc–IL-4 promoted survival in CD8+ TTE cells through upregulation of Bcl-2 and mitigation of apoptosis. Based on previous research showing that IL-4 promoted glycolysis in B cells, the researchers next evaluated whether the same might be true in T cells. Indeed, they found that in ex vivo-induced CD8+ TTE cells, Fc–IL-4 increased Glut1 expression, glucose uptake capacity, and extracellular lactate, and upon TCR stimulation, enhanced the extracellular acidification rate (ECAR); oxidative phosphorylation was not impacted. Fc–IL-4 also induced alterations in 41 metabolites, including upregulation of 3 associated with glycolysis. Analysis of metabolic genes in PMEL T cells treated with Fc-IL-4 showed enrichment for cells characterized by the glycolysis–gluconeogenesis gene module. Further, blockade of glycolysis abrogated the effects of Fc–IL-4 on CD8+ TTE cells, suggesting that promoting glycolysis is critical to treatment-enhanced antitumor immunity.
Digging deeper into the molecular mechanisms behind the changes induced by Fc–IL-4, Feng and Bai et al. performed ATACseq and transcriptome co-profiling, revealing distinct molecular profiles between treated and untreated CD8+ TTE cells, with treated cells showing changes in cytotoxicity and survival markers. Differential motif analysis of ATACseq data showed that motifs for Stat6 transcription factor binding were most enhanced, while motifs for Foxo1 transcription factor (a negative regulator of mTOR) binding were most reduced. In line with this, analysis of signaling pathways regulated by differentially expressed genes revealed that Fc–IL-4 was associated with significant upregulation of mTOR, eIF4, p70S6K, PI3K/AKT, JAK–STAT signaling, NF-κB activation, and glycolysis. Upstream regulator analysis also predicted upregulation of NF-κB, Myc, Pi3k, Akt1, and Stat6, which were found to promote glycolysis. Knockout of STAT6 partially attenuated the efficacy of Fc-IL-4 in an ACT model, and the addition of blockades for AKT or mTOR fully abrogated its benefits, pointing to a role for both STAT6 and PI3K-AKT-mTOR signaling in Fc–IL-4-mediated antitumor immunity.
To identify enzymes involved in Fc–IL-4-induced glycolysis, the researchers found that LDHA showed the most pronounced upregulation after treatment, dependent on STAT6 and PI3K-AKT-mTOR signaling, and played a critical role in Fc-IL-4-induced responses. Given that LDHA facilitates NAD+ production for glycolysis, the researchers evaluated NAD+ levels. While NAD+ was generally reduced in CD8+ TTE cells compared to progenitor exhausted T cells, treatment with Fc–IL-4 increased NAD+ in ex vivo-stimulated CD8+ TTE cells, dependent on LDHA. Increases in metabolites involved in nicotinate and nicotinamide metabolic pathways were also observed after treatment, and supplementing cells with nicotinamide riboside (a NAD+ precursor) showed similar results to Fc–IL-4, suggesting that treatment increased NAD+ generation.
Together, these results suggest that Fc–IL-4 can direct enrich and enhance the antitumor immune functions of tumor-infiltrating antigen-specific CD8+ TTE cells by binding directly to IL-4Rɑ, triggering STAT6 and PI3K-AKT-mTOR signaling. This, in turn, mediates increased glucose uptake and NAD+ concentration, which enhances glycolysis, dependent on LADH, and improves the survival and effector functions of CD8+ TTE cells. In mice, these effects enhanced antitumor immunity alone and in combination with existing ACT and ICB immunotherapies, suggesting strong potential for future use.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, co-first author Bing Feng answered our questions.

What was the most surprising finding of this study for you?
Unlike a type 1 immune response, which has traditionally been the target of cancer therapies like immune checkpoint inhibitors and CAR T cells, type 2 responses are mobilized to fight parasitic immune threats, like worms. Until now, researchers thought that type 2 immune factors were not useful in fighting cancer, and could even promote tumor growth. In this study, surprisingly, we found IL-4, a type 2 immune cytokine, could enhance current immunotherapies, which are all type 1-centric, leading to enduring antitumor immunity and long-term survival in animal models. Our results show that type 1 and type 2 immunity can be thought of in terms of synergy, like yin and yang. This discovery represents a major paradigm change in the field of cancer immunotherapy, from type 1 centric to a synergy of type 1 and type 2 immune responses, therefore is critical for designing next-generation cancer immunotherapy with improved patient responses.
Specifically, we show that IL-4 signals can potently enhance IFN-γ secretion and boost cytotoxicity of functionally impaired CD8+ T cells, particularly those in a terminally exhausted state. Another unexpected part is that STAT6, not STAT5, was crucial for IL-4's role of promoting CD8+ T cell effector function, despite IL-4 being a γc cytokine typically associated with STAT5.
What is the outlook?
Our findings suggest that IL-4 could synergize with existing cancer immunotherapies, including CAR T cell and immune checkpoint blockade (ICB) therapy. We next aim to translate this discovery to clinically applicable therapies. Two major strategies are under development. One is to use combinatory therapies involving Fc-IL-4 (a long-circulating cytokine) in combination with CAR T cells or ICBs, and the other is to develop IL-4-secreting CAR T cells. We have accumulated successful prior experience in clinical studies in IL-10-expressing CAR T cells (Nat. Biotech. 2024; Nat. Immunol. 2021), which are being tested in several ongoing first-in-human investigator-initiated clinical trials (ClinicalTrials.gov IDs: NCT06393335, NCT05715606, NCT05747157, NCT06120166, NCT06277011). The Preliminary findings have been reported in AACR 2024, EHA 2024, ASGCT 2024, SOHO 2024, etc. We are confident that the new discovery of the role of IL-4 in cancer will likely make some impacts in the clinic.
Moving forward, we are also interested in investigating other type 2 immune factors, including various cytokines and immune cells, to further develop next-generation cancer immunotherapies.
What was the coolest thing you’ve learned (about) recently outside of work?
The coolest thing is that I found my soulmate and life partner recently, and we are getting married soon…



