
This week’s extensive special feature covers select talks from the SITC - 39th Annual Meeting 2024 in Houston, TX. We have organized the content by topics below.
Keynote lecture
Rafi Ahmed
Harnessing innate immunity
Eric Vivier
Nicholas Huntington
Cancer vaccines
Elizabeth M. Jaffee
Cathy Wu
CAR T cells and T cell engagers
Yvonne Chen
Matt Maurer
Amanda V. Finck
Pablo Martinez
Fecal microbiota transplant
Arielle Elkrief
TIL therapy
George Coukos
Frank Lowery
Deciphering and modulating the tumor immune microenvironment
Jérôme Galon
Wolf H. Fridman
Aurélien Marabelle
Padmanee Sharma
Keynote lecture
What is T Cell Exhaustion? - Rafi Ahmed, PhD - Emory University School of Medicine
In recent years, research on various T cell states has developed fast, and interest in the exhausted T cell population has rapidly increased. In his keynote address, Rafi Ahmed discussed the current understanding of T cell exhaustion. Providing an overview of CD8+ T cell populations during chronic viral infection, he discussed three main subsets: (1) PD-1+TOX+TCF1+Bcl6+ stem-like cells (quiescent, with proliferative potential, resident in lymphoid tissues) that can proliferate and differentiate into (2) transitory effector PD-1+TOX+ cells (circulating cells, with reduced TCF1 expression and increased Tim3, Tbet, and Ki67 expression), and finally differentiate into (3) terminally differentiated PD-1+TOX+Blimp1+ cells (found at sites of infection, express lower levels of costimulatory molecules and higher levels of inhibitory and effector molecules). Ahmed discussed that exhaustion is an ongoing and highly regulated T cell response under conditions of chronic antigen stimulation, and defined PD1+TCF1+TOX+ stem-like CD8+ T cells as the key cells maintaining this response. This is also the population that provides the proliferative burst observed after anti-PD-1 therapy by increasing the number of transitory effector CD8+ T cells. Ahmed and his team showed that during this process, the system compensates for the accelerated differentiation by increasing the proliferation and self-renewal of the stem-like cells. Stem-like CD8+ T cells are predominantly found in T cell zones in the tumor (surrounded by cDC1 cells) where they receive TCR signaling. However, despite these signals, they do not express effector molecules and are mostly quiescent. In transcriptional analyses, the signature for the stem-like population resembled the transcriptional signature of CD4+ Tfh cells. Ahmed then discussed work evaluating virus-specific CD8+ T cells in HPV+ head and neck squamous cell carcinoma (HNSCC). HPV-specific CD8+ T cell epitopes were mapped in treatment-naive patients using a cocktail of peptides, and alongside the widely studied E6 and E7 epitopes, epitopes from the E5 and E2 proteins of the virus were also assessed. Tetramer+ TILs were directly detectable ex vivo in relatively high numbers (up 5.8% in the primary tumor, and up to 10.6% in metastatic LN), while tetramer+ T cell frequencies in the blood were below the detection limit. Interestingly, responses to both E2 (a gene required for episomal maintenance of the HPV genome in cells) and E5 (a small protein that is a potential oncogene) were predominant targets in TILs from multiple patients. HPV-specific PD1+TCF1+ stem-like CD8+ T cells were located in the tumor stroma in these patients, but not in tumor tissue, and had the proliferative capacity to differentiate into effector-like cells. The data from this study indicated that the cells required for response to PD-1 blockade are present in treatment-naive HPV+ HNSCC tumors, suggesting anti-PD1/PD-L1 therapy might be effective in this population. In an ongoing Phase 2 study, treatment-naive patients with HPV+ HNSCC are scheduled to receive two neoadjuvant doses of atezolizumab before surgery. So far, 7 patients are enrolled and the three patients that have so far completed treatment all had a pathological response to anti-PD-L1, with increased T cell infiltration and tumor size reduction. Ahmed also mentioned a new study into HPV+ cervical cancer in India, which will enroll 3 cohorts; the first asymptomatic (high-risk HPV+, no CIN), the second precancerous (hrHPV+, CIN+), and the third cancerous (hrHPV+). The focus of the work will be to longitudinally assess HPV-specific immune responses, particularly E2- and E5-specific T cell responses. Ahmed finished his keynote talk by discussing research into when during the course of a chronic viral infection stem-like CD8+ T cells are generated. PD1+TCF1+TOX+ stem-like CD8+ T cells arise within 5 days of infection, independent of whether it is an acute or chronic infection. Thus, the immune system prepares a priori for a potential chronic infection with this stem-like chronic resource CD8+ T cell. Ahmed thinks that the fundamental knowledge being obtained on T cell exhaustion should help further the development of cancer immunotherapies.
Harnessing innate immunity
Harnessing NK Cells in Cancer Therapies - Eric Vivier, DVM, PhD – Aix Marseille University
Eric Vivier discussed how NK cells can be harnessed for cancer immunotherapy both for their direct antitumor effects and by contributing to the initiation of multicellular immune responses. Various approaches can be used for NK cell-directed immunotherapy. Similar to ICB for T cells, monalizumab is a monoclonal antibody that blocks interactions between NKG2A on NK cells (and CD8+PD-1+ T cells) and HLA-E on tumor cells; it is currently in a Phase 3 clinical trial for non-small cell lung cancer. Another therapeutic option is to use NK cell engagers. Vivier discussed developments using the antibody-based NK cell engager therapeutics (ANKET) platform. A trispecific NK cell engager targeting CD123, NKp46 (most stably expressed on tumoral NK cells), and CD16 has been shown to be successful in patients with acute myeloid leukemia. Currently, another tetraspecific NK cell engager (CD20-NKCE-IL2v) is in development, which, in addition to targeting CD20, NKp46, and CD16, carries an IL-2 variant that does not interact with CD25. In a human CD20+ B cell lymphoma mouse model, CD20-NKCE-IL2v induced more efficient antitumor activity than the CD20-targeting antibody obinutuzumab, and significantly enhanced the accumulation of activated NK cells (Ncr1, Ifng, Gzmb) in the tumor bed compared to the trispecific engager (without IL-2v). When NK cell-deficient mice were challenged with human CD20-expressing tumors and treated with an injection of NK cells from WT mice plus CD20-NKCE-IL2v, major homing of the NK cells to the tumor could be observed. In non-human primates, CD20-NKCE-IL2v was shown to deplete CD20+ B cells, and did not induce cytokine release syndrome. In vitro studies with NK cells from PBMCs of healthy individuals showed that the CD20-NKCE-IL2v induced better control of human B cell lymphoma than obinutuzumab. In samples from pre-treated patients with R/R B-cell non-Hodgkin lymphoma, CD20-NKCE-IL2v induced better depletion of autologous B cells than a CD20-specific T cell engager. Finally, Vivier discussed that CD20-NKCE-IL2v induced changes in NK cell subsets, generated cytokine-induced memory-like NK cells, and activated NK cells. These activated NK cells increased expression of receptors that can interact with tumor cells, suggesting tumor antigen-agnostic NK cell immunity could be induced by the tetraspecific NK cell engager CD20-NKCE-IL2v. Therefore, NK cell engagers can induce “bystander” activity and target tumor cells that have lost the antigen against which the engager is directed.
Next-Gen Cytokine Therapies - Systemic Cytokine Drugs that Decouple Tumor Activity from Peripheral Activity - Nicholas Huntington, PhD – Monash University
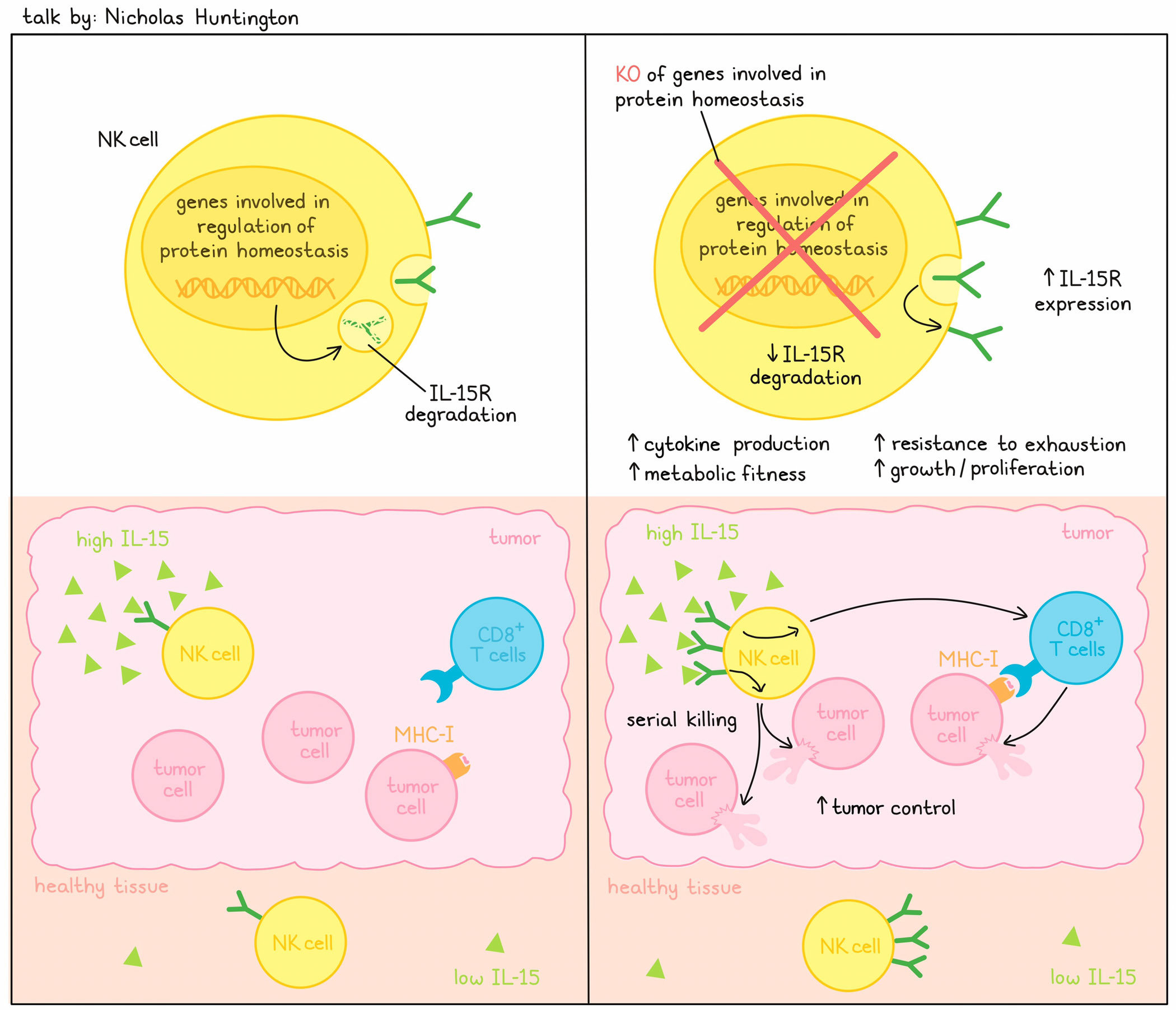
A 2008 NCI survey identified IL-15 as one of the top cytokines for potential immunotherapy, and multiple subsequent studies have demonstrated a critical role for IL-15 in multiple immune cell types. Further, direct and correlative data have linked IL-15 to good outcomes in animals and humans. Nonetheless, IL-15R agonists have failed in the clinic due to severe toxicity. Spurred on by this challenge, Nicholas Huntington focused on how to target the signaling-induced inhibitory pathway, reasoning that although IL-15R is widespread throughout the body, IL-15 levels are very low in healthy tissue (but high in tumors) and that by preventing receptor degradation, cells would be hyperresponsive to the IL-15 produced in the tumor and not activated systemically. A CRISPR knockout screen in human NK cells under conditions of low IL-15 identified a number of genes (UBE2F, ARIH2, CISH, CUL5, RNF7, DCUN1D3, UBE2L3, ELOC), whose elimination allowed better NK cell growth. All these genes were involved in protein homeostasis affecting neddylation and subsequent ubiquitination, leading to degradation. Functionally validating these results, individual knockouts of these genes in NK cells and the NK-92 cell line enhanced proliferation, cytokine production, and metabolic features of the NK cells, and the expected upregulation of IL-15R surface detection. Importantly, an inhibitor of pan-neddylation phenocopied the effects of UBE2F-gene knockouts, supporting that the pathway could be targeted for therapy. Serial cytotoxicity studies with gene knockouts in NK cells or CAR NK cells demonstrated robust serial killing, indicating the cells were metabolically very fit and resistant to exhaustion, which translated to enhanced control of MHC-I-expressing or -deficient tumors in syngeneic mouse models. The unexpected observation of activity against MHC-I-expressing tumors was a direct effect of CD8+ T cells, but enhanced by hyperactive NK-mediated improvements to the T cell response. Most of the identified gene targets were part of a degradative pathway; some could be deleted with minimal effects, and so were non-essential. Importantly, some of these targets were enzymes, setting up the opportunity to identify small molecular inhibitors specific to this class of drug targets.
Cancer vaccines
The Era of Cancer Vaccines Has Arrived - Elizabeth M. Jaffee, MD - Sidney Kimmel Cancer Center, Johns Hopkins University
Liz Jaffee, recipient of the 2024 Richard V. Smalley Memorial Award and Lectureship, provided a historical perspective on cancer immunotherapy. She emphasized the decade it had taken for immunotherapies, such as immune checkpoint blockers, CAR T cells, and TCR-engineered T cells, to be developed to help patients, and likened that to cancer vaccines, which, after years of preliminary work, are now emerging as viable therapeutic options. Jaffee’s own journey as a cancer vaccinologist began in 1990 when she collaborated with others in the development of GVAX, an autologous whole tumor cell GM-CSF-secreting vaccine, which recruits dendritic cells and induces CD4+ and CD8+ T cells. Wishing to target pancreatic cancer, a disease with a low fraction of tumor cells in the cancer mass (limiting development of autologous vaccines), Jaffee developed an allogeneic form of GVAX to target antigens shared between patients. Such vaccines identified mesothelin as a shared target, provided the first in vivo demonstration of cross-priming in humans, were immunologically active, and were associated with extended long-term survival in some cases. As pancreatic cancer presented multiple challenges, including limited T cell infiltration and a suppressive tumor immune microenvironment (TIME), Jaffee developed a two-step approach of vaccination followed by checkpoint blockade (ICB), and subsequently uncovered the need to also include co-stimulatory agonists. The neoadjuvant setting provided a window of opportunity to treat multiple sequential patient cohorts and use analysis of the resected tumors to inform the approach for the next arm. Patients were treated with vaccine alone, vaccine with anti-PD-1, or vaccine with anti-PD-1 and a CD137 agonist. Vaccine alone altered the TIME and resulted in the robust appearance of active tertiary lymphoid structures (TLSs), including T and B cells, in 33 of 39 patients. Development of TLSs correlated with better long-term survival. CYTOF and spatial profiling identified mature TLSs with recruited follicular dendritic cells, and involuted TLSs associated with sites of resolving tumor. Further, single-cell analysis identified a signature of immune activity in the tumor and in the draining lymph node, supporting both tumor and lymph nodes as sites of T cell activation. Additionally, that signature was associated with significantly improved long-term survival across cancer types. Phenotypically, the increased intratumoral immune cells were less active, but the addition of anti-PD-1 therapy enhanced signals of activity and the appearance of Th17 cells, a cell type frequently associated with clinical benefit. GVAX/anti-PD-1 led to upregulation of CD137 (4-1BB), particularly among more effector-like cells. Addition of a CD137 agonist improved tumor control in a murine pancreatic cancer model and in the clinic. GVAX plus anti-PD-1 and agonist CD137 significantly improved the phenotypes of T cells in the resected tumors. Turning to the exciting advance in the capability to target neoantigens, Jaffee described her synthetic long-peptide (SLP) vaccine trials targeting mutations in KRAS, the earliest appearing mutated oncogene in pancreatic cancer, which may open up the opportunity for early interception of cancer – a field of growing interest. In the adjuvant setting, the SLP vaccine (targeting all 6 common KRAS mutations) is delivered initially with anti-CTLA-4, and following priming with anti-CTLA-4 and anti-PD-1. Most patients responded to all 6 KRAS mutations, with significant ex vivo ELISPOT increases in both CD4+ and CD8+ cells (dominated by CD4+ T cells). Multiple TCR clonotypes could be identified, and mapping these back to bulk tumors showed that CD4+ T cells evolved to primarily a central memory phenotype, while CD8+ T cells evolved to effector memory. These TCRs were cytotoxic when engineered into Jurkat cells. Interestingly, a single TCR often recognized more than one mutated epitope, and some “public” TCRs (identical TCRs identified in different patients) were observed. In a trial in metastatic colorectal cancer, the vaccine has led to some radiographic responses. Finally, to close, Jaffee described her efforts toward even earlier interception by characterizing the immune state of pancreatic intraepithelial neoplasia (PanINs). She found that PanINs harbored immature TLSs containing T and B cells, but not follicular dendritic cells, and Jaffee suggested that vaccines would be the ideal candidate to further mature such structures. Early results from a clinical trial in patients with PanINs who were at a high risk of developing pancreatic cancer showed that most patients generated responses to all 6 KRAS mutations contained within the vaccine.In two patients with 1 year follow-up, responses were durable. In another ongoing trial in patients with intraductal pancreatic mucinous neoplasm (IPMN) at high risk for malignant transformation, the observable cysts may be amenable to future resection and analysis.
Human Neoantigen-Specific CD4 T Cells in the Native Setting and Following Cancer Vaccination - Cathy Wu, MD - Dana-Farber Cancer Institute
In a session devoted to the evolving and important role of CD4+ T cells in antitumor immunity, Cathy Wu presented her results on the characterization of CD4+ T cells in the context of vaccination. Beginning with a short history of the key technologies that led to the emergence of neoantigens as vaccine targets based on their exquisite tumor specificity and expected high immunogenicity, Wu noted that across various vaccine platforms, CD4+ T cells are the dominant induced cell type, despite selecting epitopes based on MHC-I binding. This led to the initiation of a program to understand the baseline and vaccine-induced profile of CD4+ T cells using single-cell sequencing and TCR reconstruction technologies that allowed interrogation of hundreds of antigens and TCRs simultaneously. The focus of this research was on melanoma, which expresses MHC-II in inflamed tumors and occasionally in non-inflamed tumors. Wu’s published work in the adjuvant melanoma setting showed that prior to vaccination, CD4+ T cells could be found in a memory state (rarely tumor antigen-specific), an exhausted state (mostly tumor antigen-specific), a follicular helper/precursor exhausted state (moderately tumor antigen-specific) and a regulatory state (strongly tumor antigen-specific). Interestingly, tumor-reactive Tregs were found only in the patients whose tumors expressed MHC-II, and were most strongly induced in patients with a high mutation burden, suggesting an immune-dampening effect towards induced CD8+ T cells as a possible tumor escape mechanism. Vaccination with neoantigen vaccines led to strong CD4+ T cell responses and weaker CD8+ T cell responses in patients with melanoma, both of which persisted for years. Production of CD4+ tetramers allowed identification of a clear differentiation pathway (naive to memory) over time and the demonstration that a broad repertoire of TCRs was induced. Importantly, all the patients showed durable clinical benefit (absence of recurrence or recurrence controllable with additional therapy), including two patients with recurrence who demonstrated enduring CRs after subsequent treatment with anti-PD-1 therapy. Wu closed by describing new results from a recently conducted melanoma trial including patients with high-risk, unresectable stage IV disease, treated with a newly formulated peptide vaccine (including the well known adjuvant Montanide and poly IC:LC) and local anti-CTLA-4. In this trial, patients were treated with anti-PD-1 therapy while the vaccine was prepared. Very strong CD4+ responses were detected, but also, the strength of CD8+ responses was strongly increased compared with results from the previous clinical trial. Bulk TCR sequencing of peripheral blood showed a wave of induced TCRs after the anti-PD-1, and a second wave of different TCRs after vaccination. Phenotypic analysis of TILs from patients with existing or progressive disease suggested that disease control was associated with an increase in CD8+ T cells and a decrease in CD4+ Tregs, and with effector-like CD4+ T cells that appear less exhausted, pointing to a potentially important role in disease control.
CAR T cells and T cell engagers
Multi-Pronged CAR-T Cell Therapy for Cancer - Yvonne Chen, PhD - University of California, Los Angeles
Discussing structural considerations for CAR engineering, Yvonne Chen began her talk by reviewing the multiple structural and logic-driven CAR options, but cautioned that “simpler is better” in regards to manufacturing considerations. Details do matter though, and even subtle differences in the very basic components of CARs can result in very different outcomes. First, although many types of binding domains can be utilized (antibodies, nanobodies, natural ligands, in silico-designed molecules), seemingly minor differences in the binder sequence can dramatically alter CAR signaling behavior. Studying why CD20 CARs do not function as well as CD19 CARs, Chen and colleagues built two CD20-CARs using scFvs that were 91% identical in sequence, but behaved completely differently in terms of tonic signaling (how strongly the CAR signals in absence of antigen), with one showing no tonic signaling and the other showing very high tonic signaling. Generally, tonic signaling is considered a bad characteristic, as it drives premature exhaustion, but the CAR without tonic signaling functioned poorly in vivo. Therefore, the lab hypothesized that a little tonic signaling is needed for best functioning, similar to TCRs, which do show a low level of tonic signaling. Using a hybridization strategy combining the CDR regions of one antibody with the framework of the other, they found that the hybrid functioned better than either of the original CARs, and even better than the CD19 CAR. To further investigate why the CARs had tonic signaling, Chen looked into the signaling domains. It is well known that there are multiple different properties associated with CD28 vs 4-1BB in CAR constructs, and tonic signaling of CD28 has a bad reputation. Assessing whether CD28 was indeed the culprit by creating various chimeric CARs, they found that CD3ζ, and not CD28, was the main contributor to tonic signaling (as well as to antigen-independent proliferation and multiple metabolic characteristics), although that was still not the complete story. Chen also found that the protein conformation, in this case the relative angle of rotation between the ecto- and endodomains, also impacted tonic signaling. Using alanine additions to the base of the transmembrane domain to extend the domain helix, different twists in the CAR molecule were created, which dramatically altered tonic signaling and impacted T cells’ proliferation and ability to control tumors in vivo. Finally, Chen highlighted some practical considerations for CAR implementation by discussing the importance of the transgene payload size and transduction efficiency (impacting the CAR expression on a cell), transgene stability, and the stoichiometry of different components, which have potential impacts on cell expansion ex vivo and post-infusion.
Preclinical development of MT-303, a novel LNP-Formulated GPC3-Specific CAR mRNA, for in vivo programming of monocytes to treat hepatocellular carcinoma - Matt Maurer, MD – Myeloid Therapeutics
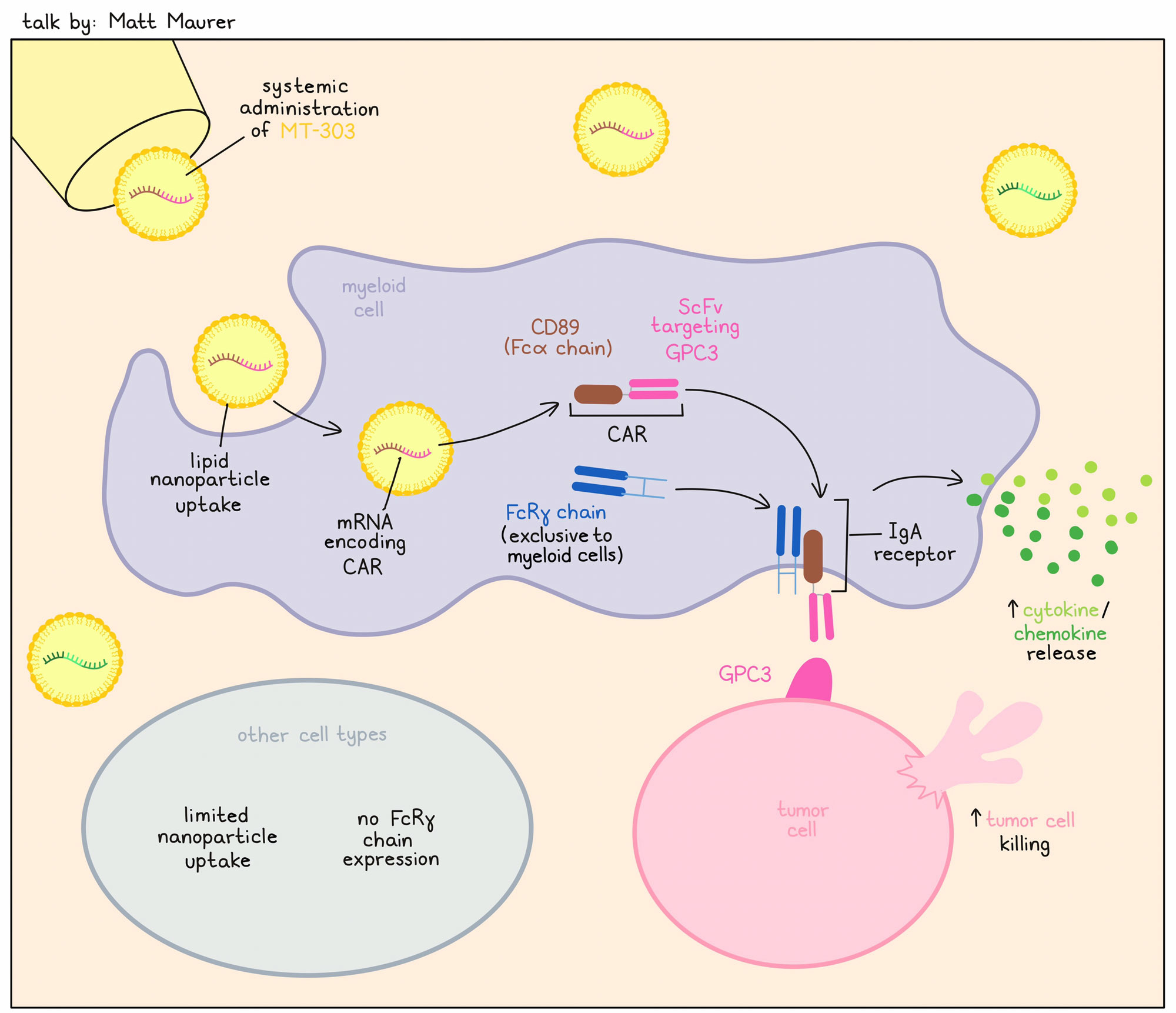
Matt Maurer discussed Myeloid Therapeutics’ work on myeloid cell programming for immunotherapy. Myeloid cells are foundational to control of pathogens, and can easily penetrate the tumor microenvironment, but generally do not act on tumor cells. The focus of Myeloid Therapeutics has been to reprogram myeloid cells in vivo to recognize the tumor, killing and phagocytosing target cells and activating NK and T cells. Reprogramming is accomplished by using mRNA coding for a CAR, which is formulated into lipid nanoparticles (LNP) and delivered intravenously. Such an off-the-shelf therapy could enable antitumor responses without the lengthy manufacturing process that other CAR therapies require. MT-303 is an LNP-encapsulated GPC3-targeting CAR that specifically induces stable CAR expression and activity in myeloid cells only. The mRNA encapsulated in LNP produces a chimeric molecule consisting of an scFv targeting GPC3 linked to CD89. CD89 is the Fcα chain, which specifically associates with the FcRγ chain, present only in myeloid cells (the Fcα:FcRγ heterodimer is the IgA receptor). The restricted expression of FcRγ to myeloid cells provides the unique specificity following in vivo delivery. Preclinical research optimized the sequence of the coding region and the untranslated regions, which enhanced GPC3 CAR expression levels and improved the durability of CAR expression. Electroporation of the GPC3-encoding CAR to PBMC-derived monocytes resulted in GPC3 CAR-expressing monocytes that killed tumor cells and released chemokines and cytokines in vitro. Among PBMCs, only monocytes took up GPC3-CAR (LNP) and induced CAR expression. Robust CAR expression on monocytes and chemokine/cytokine release was also observed in tumor-bearing mice treated intravenously with MT-303. Dose-dependent efficacy of MT-303 was also observed in a GPC3+ hepatocellular carcinoma (HCC) xenograft model. As this work was performed in NCG mice, only the direct effects of monocytes were observed. Final preclinical safety analyses were performed in cynomolgus monkeys, in which a dose-dependent GPC3 CAR expression could be observed in the peripheral blood in monocytes only. Two human trials are ongoing, including a recently started MT-303 study in HCC and other cancers expressing GPC3. Further, another product, MT-302, which induces a TROP2-targeting CAR on monocytes, is being assessed in a basket study against tumor types with known high TROP2 expression. The MT-302 clinical trial has so far shown that the therapy increases cytokines and chemokines, induces CAR expression, and increases T cell responses (induction of novel TCR clonotypes and increased clonality). Antitumor responses have been detected in biopsies. So far, both products have been well tolerated with repeat dosing (no dose-limiting toxicities) without the need for preconditioning.
Ping-pong Dual CAR-T Cells: boosting in the periphery to enhance antitumor efficacy in solid tumors - Amanda V. Finck, BS – University of Pennsylvania
Amanda Finck discussed how CAR T cells directed at solid tumors often fail to persist in the periphery, which is a problem, as persistence has been associated with better outcomes. One approach to overcome this is to increase the dosage of CAR T cells infused. However, when large numbers of CAR T cells are administered, they localize in the lung immediately after infusion, resulting in the recognition of low levels of antigen, which induces toxicity (first pass hypothesis). To overcome this issue, Finck hypothesized that dual CAR T cells with the capacity to engage with antigen in the periphery might be used, allowing for lower initial dosing to prevent the first-pass toxicity, but with later boosting upon engagement with antigen in the periphery, increasing their expansion and persistence. To investigate this, Finck and colleagues used CD19 as the secondary antigen in the periphery, as B cell depletion can be safe. Using 1 million CAR T cells in a syngeneic model without lymphodepletion, B cell aplasia was observed by day 5, and there was a relapse in the B cell population by day 26. At both time points, there was a boost in the periphery in the overall number of CAR T cells. In a PDAC model, the dual CAR T cells improved tumor control and overall survival, without any increase in toxicity based on tissue analysis following necropsy. In the spleen, similar to the periphery, B cell aplasia was observed, and there was a boost in CAR T cells. Once B cells repopulated over time, there was also another spike in the number of CAR T cells. Importantly, in the tumor, there were higher numbers of these CAR T cells at all time points, and they were more highly activated (increased % of PD-1+, Lag3+, or CD39+). To model this with human cells, NSG mice were used, but as these do not have B cells, mice were first injected with donor-matched B cells before treatment with the CAR T cells. In this model, the same increase in CAR T cells was observed, as was control of tumor burden, without aberrant toxicity.
Tarlatamab: a Bispecific T-cell Engager Immunotherapy Paving the Way for Small Cell Lung Cancer and Solid Tumors - Pablo Martinez, MD, PhD - Amgen
Pablo Martinez presented translational and clinical data on tarlatamab, the just recently approved T cell engager for the treatment of patients with second-line extensive-stage small cell lung cancer (SCLC). Tarlatamab’s target antigen delta-like ligand 3 (DLL3) is a regulator of the Notch pathway, and is usually expressed intracellularly, but is uniquely expressed on the cell surface in neuroendocrine tumors, including SCLC. For the development of tarlatamab, Martinez and his team built on the knowledge and experience gained with the CD19-specific T cell engager blinatumomab. Blinatumomab has a short half-life (2-3 hours) and requires continuous i.v. infusion over 28 days, and so the half-life of tarlatamab was extended by the addition of an Fc-proportion, allowing for a more comfortable administration schedule of every two weeks. A key clinical challenge with T cell engagers is identifying the dose, particularly during the initial phase, when step up doses are utilized to mitigate the risk of severe cytokine release syndrome (CRS). The early drug development of tarlatamab required a lengthy and complex dose exploration and expansion phase I study, which was open for 6 years and enrolled more than 100 patients. Part 1 of the phase 2 trial was a dose optimization study that demonstrated similar response rates between the 10 mg and 100 mg dose cohorts, but a better safety profile with almost no grade 3 or higher CRS and very limited ICANS in the 10 mg dose cohort. CRS was largely confined to the first or second dose. Accelerated approval was granted based on the phase 2 trial, and the confirmatory phase 3 trial is still ongoing. With lessons learned from blinatumomab, Martinez and his team are interested in studying tarlatamab in earlier lines of disease after tumor debulking, and several studies are currently enrolling patients with first-line or limited-stage SCLC and in combination with anti-PD-1/L1. Tarlatamab is currently under investigation in other DLL3+ neuroendocrine tumors. More than 95% of SCLC tumors demonstrated surface expression of DLL3. In some other tumor types, DLL3 expression is less common, and may require selection for inclusion in clinical trials. Finally, Martinez pointed out that Amgen has more T cell engagers with novel designs in the pipeline.
Fecal microbiota transplant
Phase II Clinical Trial of Fecal Microbiota Transplantation in Combination with Immunotherapy in Patients with Lung Cancer and Melanoma (FMT-LUMINate) - Arielle Elkrief, MD - University of Montréal Research Center
Fecal microbiota transplantation (FMT) has become an interesting way to modulate the gut microbiome to improve clinical responses to immune checkpoint blockade (ICB). Arielle Elkrief discussed a Phase 2 trial in which FMT from healthy donors was given to patients a week before they received standard-of-care first-line ICB. The trial consisted of several cohorts, including one cohort of patients with PD-L1hi non-small cell lung cancer (NSCLC) treated with pembrolizumab, a second cohort of patients with cutaneous melanoma treated with ipilimumab and nivolumab, and a third small exploratory cohort of patients with uveal melanoma. In the NSCLC cohort, the overall response rate (ORR) was 75%, with 15 patients having a durable partial response (PR). In the cutaneous melanoma cohort, an ORR of 75% was observed (five patients with complete responses and 10 patients with PRs). Both ORRs were higher than historical data. In NSCLC, no grade ≥3 toxicities were observed, suggesting FMT might have been decreasing immune-related adverse events (irAEs) in this population. In cutaneous melanoma, an incidence of grade ≥3 irAEs of 65% was observed, including three patients who experienced myocarditis. The toxicity observed occurred earlier compared to historic trials with ipilimumab and nivolumab. When assessing donor-specific effects on toxicity, many of the grade ≥3 irAEs were observed in those treated with a specific donor sample (donor 5). Preliminary metagenomics data showed that pre-treatment healthy donor microbiomes clustered separately from that of the patients, and non-responders and responders overlapped. However, there was a separate clustering for the microbiome of responders and non-responders detected after FMT. Notably, donor 5 clustered separately from the other healthy donors. In post-FMT samples, those with irAEs or colitis had a decrease in bacterial richness. Further, the leaky gut marker sST2 was decreased in responders post-transfer, but increased over time in those treated with ipilimumab and nivolumab, and was associated with grade ≥3 irAEs. When assessing PBMCs, there was an increase in activated central memory CD4+ T cells and intestinal CD4+ T cells in responders post-FMT. To further confirm these data, murine experiments were conducted under pathogen-free conditions. Recapitulating the clinical trial, mice received FMT from patients, followed by FMT from matching donors, and were then inoculated with tumors and treated with PD-1 and CTLA-4 ICB. This confirmed that the donor FMT was responsible for the tumor control and therapeutic activity. Using samples from a responder and a non-responder who both received FMT from donor 5 revealed an increase in bacterial richness in the responder, which was not observed in the non-responder. Further trials are ongoing to assess longitudinal effects on the microbiome in patients during treatment to better understand the compatibility between donor and host, and how to best stratify patients for therapy.
TIL therapy
TIL Therapy: What matters? Lessons from a deep dive - George Coukos, MD, PhD - Ludwig Cancer Research Lausanne Branch
While it has been clinically established (and approved as a therapy in the US and some EU countries) that adoptive cell transfer of tumor-infiltrating lymphocytes (TIL-ACT) can be effective in melanoma, only about 36% of patients respond. To understand why some patients respond and to increase the response rate, George Coukos discussed three lessons learned from research into the differences between responders and non-responders in a clinical TIL-ACT study in 13 patients (6 responders [R; CR or PR]; and 7 non-responders [NR; SD or PD]) with melanoma in whom PD-1 therapy had ultimately failed. The first lesson learned was that more tumor-resident cells in the product resulted in better outcomes. Responders had higher numbers of total cells in the infusion product (all Rs received at least 50 x 109 cells). TCR sequencing demonstrated that a higher fraction of these cells were tumor-resident, and a large fraction of these were tumor-reactive TILs. Baseline tumors had a large representation of tumor-reactive TCRs, but interestingly, the infused product lost about two-thirds of these clonotypes, and even fewer were found in day 30 biopsies (of a separate tumor lesion). How to improve this limited perseverance could be an important advance. A second lesson was that the majority of tumor-reactive TILs in the infusion product were derived from the exhaustion compartment (either precursor or permanently exhausted), and had signatures of tumor or neoantigen reactivity. Responding patients’ tumors harbored activated TILs at baseline. The CD8+ TIL compartment was significantly enriched for cells expressing signatures of exhaustion, TCR signaling, cytotoxicity, and IL-2 signaling at baseline. Importantly, precursor-exhausted T cells were found to have the highest expansion rate. Finally, the third lesson was that exhausted TILs required myeloid niches to function properly and respond to IL-2 in culture. Tumors of responders exhibited dense inferred TIL:DC crosstalk by RNAseq at baseline in tumors and stroma, which was not observed in NRs, and IHC demonstrated closer physical distances between CD8+ and CD11c+ cells in both the tumor and the stroma. Coukos and team showed that synapses formed between DCs and CD8+ T cells. Further, responders had higher CD28 signature scores, and PD-1+CTLA-4+CD137+CD8+ TILs expressing CD28 had higher levels of pSTAT5, IL-2, and Ki67. These data suggest that interactions with DCs are needed for T cells to maintain responsiveness to IL-2 and persevere. However, when myeloid cells produce prostaglandin E2 (PGE2), they become suppressors. Exposure to PGE2 induces the loss of IL-2Rγ in TILs, and the subsequent reduction in IL-2 signaling leads to T cell dysfunction and reduced TIL expansion.
Tumor neoantigen reactivity-selected TIL for the treatment of metastatic gastrointestinal cancers - Frank Lowery, PhD – National Cancer Institute
After observing a response rate of ~ 50% and long-tail progression-free survival of ~20% in patients with metastatic melanoma treated with TIL therapy, Frank Lowery and colleagues were interested in developing TIL therapy for epithelial cancers. TILs can be expanded from metastatic gastrointestinal (GI) tumors, but with a lower proportion of CD8+ T cells than observed for melanoma. Treatment of 18 patients with GI cancers with bulk, unselected TILs led to tumor shrinkage of more than 30% in two patients at a single follow-up, but no confirmed RECIST 1.0 responses were seen. To improve the efficacy of the therapy, Lowery and colleagues emulated an earlier observation in a patient with cholangiosarcoma who was successfully treated with a TIL fragment enriched for a neoantigen-specific TIL population. TIL fragments were screened against tumor-encoded candidate neoepitopes, and positive reactive cultures were expanded for subsequent treatment of the patient. A previous study showed that neoantigen-reactive TILs could be found in 83% of patients with GI cancers. In a clinical trial, 3 of 39 patients with GI cancer who were treated with selected TILs achieved a confirmed partial response (PR), with one long-term PR still ongoing after more than 6 years. To further improve this neoantigen-selected TIL therapy approach, treatment was combined with pembrolizumab (anti-PD-1). In this trial, more than half of the patients had shrinkage of their tumor at the first follow-up. Unfortunately, these responses were not durable, and only 8 of 34 patients had a confirmed PR on this trial. Retrospective analysis confirmed that across samples, ~35% of the infused TIL product was neoantigen target-reactive. A high number of neoantigen-reactive CD4+ T cells, but not CD8+ T cells, corresponded with response, and about 2.5 x 1010 reactive CD4+ T cells were infused within the 8.5 x 1010 TIL product. Infusion products of responding patients targeted about 1.5-fold more distinct neoantigens than non-responding patients, and in tumor resections, 95% of the targets appeared to be clonal. Target mutations were mainly patient-unique, and 82% were expressed (RNAseq). However, most of the neoantigen-reactive TIL from metastatic GI cancers were in a dysfunctional state, and 85% were in a hyper-exhausted cluster (Tox+, CD39+, PD-1+, TCF7-, IL7R-). Comparison of the infusion products in this GI trial with bulk TILs of patients with melanoma who had a complete response in earlier trials revealed 10x fewer CD8+CD39-CD69- TILs in GI TIL products. Higher numbers of CD39-CD69- TILs corresponded best with response in the melanoma trial. While transcriptomic phenotyping is still ongoing, Lowery and colleagues observed more TCF+ TIL than anticipated. Bulk RNAseq of tumors resected for TIL therapy production may already inform about response, with “hot” versus “cold” tumor features seeming to predict response.
Deciphering and modulating the tumor immune microenvironment
Immune Contexture of Premalignant Lesions - Jérôme Galon, PhD - INSERM, Sorbonne Université Paris
As a testament to the importance of the immune system in cancer, analysis of the state of immune cells (immunoscore; the type, density, and location of immune cells) and immune cell quality (Immunosign) have proven to be the most effective prognostic markers for patient outcomes, including metastasis and survival, and reflect the ongoing co-evolution of tumors and immune responses. Jérôme Galon, a pioneer in the field of analysis of the tumor immune microenvironment, presented his results extending this analysis to the premalignant setting, including observations that even at the earliest stage of tumor development (T1), T cell priming was already occurring. Premalignant lesions have been classified to date into multiple stages of abnormal expansion (“...plasia”). Dysplasia is the progression from normal tissue to carcinoma in situ. Bronchoscopy biopsies from a large cohort of heavy smokers provided samples for each of these premalignant stages for in-depth analysis. Immune cell infiltration increased across the tumor development spectrum, and transcriptomic analysis revealed clear RNA expression changes and patterns, with evidence that immune evasion was already occurring before “invasion” – the marker of cancer. The pattern of certain transcriptomic increases occurring late and strongly during pre-malignant progression (“ascending from high grade”) was particularly rich in immune signatures, including both adaptive and innate immune cells. Functionally, T cells were observed to be shifting from naive to memory states, with similar functional changes noted for B cells and mast cells. Gene expression for immune checkpoints, costimulatory molecules, cytokines were similarly shifted, again reflecting increased immune evasion, even in premalignant lesions. Pre-malignant colonoscopy screening samples from a large cohort of patients provided the opportunity to conduct longitudinal analysis. Patients were separated into groups in which polyps did not recur (F1) and those in which polyps or cancer did recur (F2 and F3 [with higher rate]) during a follow-up period of up to 10 years. Single-cell sequencing could clearly track the transition through different pre-cancerous states to actual carcinoma, identifying distinct patterns of gene expression for each state. There was no clear association between the pre-cancer development rate or recurrence and the major histological subtypes (adenomatous or serrated), the molecular pathways (CMS1-4 classification), or the pattern of mutated oncogenes (although gene mutations did very clearly distinguish the histological subtypes). Lesions from non-recurring patients did, however, have higher immune cell infiltration, and spatial analysis by multiplex IHC detailed both the cell types and interactive patterns (formation, number, and maturity of tertiary lymphoid structures) associated with these non-recurring high gene profile samples. This detailed understanding of how the immune system responds to developing tumors may reveal new therapeutic options to prevent progression from the pre-neoplastic state, dramatically reducing morbidity and mortality in patients with cancer.
Tumor Cell Heterogeneity Governs Spatial Organization of the Tumor Microenvironment - Wolf H. Fridman, MD, PHD - Cordeliers Research Centre
Tertiary lymphoid structures (TLSs) in tumors have been associated with better outcomes and immunotherapy responses. Wolf Fridman discussed research on how TLSs modulate these effects. Spatial research showed areas of B cell-rich hotspots in TLS-positive tumors. In TLSs, naive and memory B cells, as well as plasmablasts and plasma cells were detected, suggesting B cells may mature to plasma cells in TLSs. Expression of TLS RNAseq signatures was associated with selection and amplification of B cell receptor (BCR) clones. Further, more B cell clonotypes and clonal amplification were present in tumors with TLSs. These tumors also had increased immunoglobulin (Ig) production, and Ig genes were highly expressed inside and at a distance from the TLSs. TLS-positive tumors had increased repertoire diversity for most IGH clonotypes, and the same clonotype for IgG and IgA was detected within a tumor, suggesting clonal switching was taking place locally. Spatial research showed switched plasma cells producing different isotypes to migrate into distinct tumor compartments from the TLSs. IgG-labeled tumor cells were enriched in tumors highly infiltrated with IgG+MUM1+ plasma cells, and these IgG antibodies could induce macrophage-dependent apoptosis of tumor cells. In patients with renal cell carcinoma treated with ICB, response rates and outcomes were much higher in tumors with high levels of IgG. These types of B cell responses might explain how some tumors with low mutational burden respond to immunotherapy. Tumor-specific IGH clones could be detected in the blood of patients, and current research is focused on determining whether these correlate to therapy response, and whether they could potentially be used as a therapeutic. Finally, Fridman discussed the impact of intratumoral heterogeneity on TLS spatial distribution by assessing dedifferentiated liposarcoma. This cancer type contains a mixture of regions with various levels of differentiation, allowing for study of various TMEs within the same tumor. In these tumors, the number of TLSs correlated with the relative area of high- and low-grade dedifferentiated regions. Spatial transcriptomics showed that the low-grade areas highly expressed B cell gene signatures, while high-grade regions expressed fibroblast, mesenchymal stem cell, and trophoblast, but not immune-related signatures. Therefore, dedifferentiation may result in tumor escape from immune surveillance through increases in stromal barriers and immune exclusion.
Targeting Intratumoral Tumor-Specific Regulatory CD4 T Cells - Aurélien Marabelle, MD, PhD – Gustave Roussy
To unravel the distinct linkage, or lack thereof, between toxicity and tumor control for anti-CTLA-4 and anti-PD-1 therapies, respectively, Aurélien Marabelle and his team used multiparameter flow cytometry and scRNAseq on freshly resected human primary tumors of various histological types to directly study the effects of anti-PD-1 or CTLA-4 on intratumoral immune subsets. While PD-1 was expressed on CD4+ and CD8+ effector T cells and Tregs with a high inter-individual variability, CTLA-4 was mainly expressed on CD4+Foxp3+ Tregs. Highly expanding tumor-reactive Treg clones in the tumor were characterized by high expression levels of CD39, CD25, and CLTA-4, omitting the need for intracellular Foxp3 staining. Marabelle obtained biopsy samples from the NIVIPIT trial in which 60 patients with first-line metastatic melanoma were treated with anti-CTLA-4 (i.v. or i.t.) and anti-PD-1, and separated them according to durable clinical benefit (DCB; defined as SD, PR, or CR maintained at 6 months) or no DCB. Patients with DCB had a trend towards higher tumoral HLA-I and HLA-II expression, significantly higher numbers of PD-1+CD8+ T cells and Th1 CD4+ T cells, higher levels of secreted granzymes, and increased B cell contexture (CD20, B and plasma B cell RNA markers). Surprisingly, patients with DCB had more clonally expanded activated CD25+CD39+ Tregs in their tumor at baseline, and only in patients with DCB were these activated Tregs depleted (monitored at week 3 during treatment), with a significant increase in the tumor-reactive CD8+CD39+/CD4+CD39+CD25+ ratio. CD25+CD39+ Tregs in patients with DCB or no DCB expressed similarly high levels of CTLA-4 prior to therapy. Investigation into why treatment with anti-CTLA-4 only led to Treg depletion in some patients showed that patients with DCB had higher activating FcγR expression and higher densities of M2 macrophages at baseline, and these M2 macrophages increased even more during treatment. Marabelle hypothesized that these traditionally considered “bad biomarkers” are actually a sign of ongoing immunoediting, and are required for the efficacy of checkpoint blockade with FcγR-dependent Treg killing by M2 macrophages. Data from other trials confirm that patients with early disease stages with still active immune editing benefit more from anti-CTLA-4 treatment than patients with more advanced stages of disease.
From the Clinic to the Lab: Investigating Mechanisms of Response and Resistance to Immune Checkpoint Therapy - Padmanee Sharma, MD, PhD - The University of Texas MD Anderson Cancer Center
To gain a deeper understanding of the role of the tumor immune microenvironment on response or resistance to checkpoint blockade (ICB), Padmanee Sharma led one of the earliest teams to analyze patient tissues obtained in neoadjuvant clinical trials. Samples from patients with localized bladder cancer after pre-surgical treatment with anti-CTLA-4 plus anti-PD-L1 revealed that the responders on the trial had a higher density of tertiary lymphoid structures (TLSs) with higher numbers of ICOS-expressing CD4+ T cells. Codex analyses of the TLSs confirmed a close proximity of ICOS+CD4+ T cells, CD8+ T cells, and BATF3+ DCs. Focusing on mechanisms of resistance to ICB in tumors with low mutational burden, Sharma showed how neoadjuvant anti-CTLA-4 treatment turned a cold prostate cancer microenvironment hot by T cell infiltration, but that compensatory inhibitory pathways evolved in longitudinal samples. In particular, the percentage of VISTA+CD68+ and PD-L1+CD68+ immunosuppressive macrophage subsets increased in patients. Using a similar approach in pancreatic cancer, Sharma and her team discovered another mechanism underlying resistance to ICB. The researchers identified TSG6+ cancer-associated fibroblasts (CAFs) to be uniquely present in pancreatic tumor tissue when compared to normal pancreatic tissue by scRNAseq. TSG6 is an anti-inflammatory protein that has been previously described to polarize macrophages towards an M2-like phenotype in a wound healing setting. A closer look at TME profiles in murine melanoma (B16F10) and pancreatic cancer (mT4) revealed that pancreatic tumors demonstrated significantly higher frequencies of TSG6+ CAFs. Treatment of mT4 tumors with combination ICB and a blocking antibody to TSG6 led to increased survival, an increase in CD8+ T cells, and a decrease in suppressive VISTA+ macrophages in the TME. Sharma and her team hypothesized that the organ-specific microenvironment would need to be considered, and used scRNAseq and CYTOF to identify specific myeloid cell subsets present in glioblastoma, but not in non-small cell lung, renal, colorectal, or pancreatic cancer. The researchers singled out a CD73+CD68+ myeloid cell subset that persisted after anti-PD-1 treatment and was associated with microvascular proliferation in immunofluorescence histology of human GBM samples. Further, scRNAseq analysis revealed that this CD73+CD68+ myeloid cell subset was a signature of immunosuppression and hypoxia. In murine GBM models, treatment with anti-PD-1 and CTLA-4 improved tumor rejection and survival in CD73-knockout mice compared to wild-type mice. Sharma sees the future of ICB in combination strategies, with still many complex open questions, such as the real-time dynamic effect of treatment, scheduling of each therapy, and unwrapping the distinct subsets of each immune cell population.
By Maartje Wouters, Lauren Hitchings, Ed Fritsch, and Ute Burkhardt



