
The ACIR team attended the newly established AACR IO meeting held on February 23-26, 2025 in Los Angeles, CA, USA. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Welcome Keynote Addresses
Jim Allison
Arlene Sharpe
Neoantigens and ERVs
Catherine Wu
Christopher Klebanoff
Alena Gros
Sebastian Amigorena
George Kassiotis
Cytokines
Christopher Garcia (Keynote speaker)
CARs
Crystal Mackall (Keynote speaker)
Saar Gill
T cell Responses
Annette Paschen
Caroline Robert
Daniela Thommen
Andrea Schietinger
Tumor Immune Microenvironment
Florent Ginhoux
Jessica Stark (Keynote speaker)
In Situ Approaches
Ignacio Melero
Zhijian James Chen
Welcome Keynote Addresses
Immune Checkpoint blockade in cancer immunotherapy: Distinct biologies of CTLA-4 and PD-1 blockade-James P. Allison, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA
Jim Allison, who has been attending AACR meetings since the 1970s, was excited in his keynote address to be speaking at this inaugural conference dedicated entirely to immune oncology. He reviewed a brief history of the field, from the discovery of the TCR and CD28 costimulation, to the role of CTLA-4 and its binding partners, the B7 ligands, and to the use of anti-CTLA-4 to disable the brakes on the immune system and unleash its powers for antitumor immunity, overcoming the head-start advantage of the tumor. Since its approval by the FDA in 2011, anti-CTLA-4 has been used to treat many patients, with some seeing remarkable and durable responses. This includes a patient named Sharon, who was one of the earliest remarkable responders, and who is still disease-free today, over 21 years later. Co-targeting of PD-1 – another immune checkpoint with a distinct mechanism of action – has further improved responses in some patients, with the combination showing synergistic efficacy, and with 52% of melanoma patients still alive after 10 years. In more recent work, researchers found that anti-CTLA-4 lowers the threshold for activation (allowing cells with lower avidity to be activated), expands the TCR repertoire, strengthens T cell memory, enhances tumor infiltration, and may induce non-canonical T cell phenotypes that do not arise under natural conditions. Notably, treatment with anti-CTLA-4 leads to an increase in Th1-like CD4+ T cells expressing ICOS – a member of the CTLA-4/CD28 superfamily. In mice, ICOS agonism in the context of anti-CTLA-4 treatment increased antitumor efficacy and improved survival rates, with evidence of increased effector CD4+ T cells, increased effector memory CD8+ T cells, reduced exhausted T cells, and a shift in the myeloid cell population from more suppressive to more inflammatory, with myeloid cells actually contributing to antitumor efficacy. Looking within the TME, researchers found that cDC1s were required for the efficacy of anti-CTLA-4, dependent on ICOSL, and that they colocalized with ICOS+ CD4+ and CD8+ T cells, and with macrophages, forming tetrads. Based on these results, Allison described a proposed mechanism, so far seen only with anti-CTLA-4, and not anti-PD-1, in which ICOS+ CD4+ T cells interact with and activate cDC1s, which release IL-15. This, in turn, increases ICAM-1 expression on DCs and macrophages, stabilizing tetrads that support optimal activation of cytotoxic T cells. Finally, Allison highlighted a wide range of possible immune target molecules with potential to improve antitumor immunity either alone or in the context of novel settings that arise with other immunotherapies. While the field has made massive strides in recent years, Allison stressed the continued goal of flattening the curve for all patients with cancer, ending with a statement that “we’ve got a lot of work to do.”
The biology behind PD-1 checkpoint blockade - Arlene H. Sharpe, Harvard Medical School, Boston, Massachusetts, USA
To complement the opening keynote address on the biology of CTLA-4, Arlene Sharpe focused on the biology behind PD-1 blockade. PD-1 plays a key role regulating the TCR/CD28 signaling pathway. Upon binding its ligands (PD-L1 or PD-L2), a series of intracellular phosphorylation events result in downregulation of T cell responses, protecting tissues from inflammatory damage. Moreover, as PD-L1 also interacts with B7.1, a key ligand of CTLA-4, these components all participate in a broad network regulating T cell activity. Somewhat paradoxically, the negative signaling through PD-1 is necessary for effective memory CD8+ T cell generation. PD-1 has emerged as both a key marker and a modulator of T cell differentiation and modulates exhaustion pathways, preventing terminal exhaustion. To improve the clinical response to anti-PD-1 therapies, it is important to fully understand the biology of PD-1 in order to effectively identify other relevant co-therapies. For example, PD-1 also limits the activity of Tregs, suggesting that bispecifics that target anti-PD-1 to effector cells, but not regulatory cells, may be valuable. The ratio of PD-1+ Tregs to PD-1+ CD8+ effector T cells may be a valuable biomarker. The microbiome has also been shown to have an important role in immunotherapy response and resistance, and interestingly, germ-free mice do not respond to anti-PD-1/L1 blockade, and highly express PD-L2 on CD11c+CD11b+ cells, suggesting that this was a suppressive pathway amenable to therapy. PD-L2 binds not only to PD-1, but also to Repulsive Guidance Molecule b (RGMb), and specific blockade of the PD-L2/RMGb pathway restored antitumor activity in germ-free mice. Evaluation of stool microbiota transplanted from patients who did or did not respond to anti-PD-1 therapy revealed that non-responder microbiota upregulated PD-L2 and RMGb. Notably, blocking the PD-L1/RMGb pathway restored the antitumor activity of anti-PD-L1, highlighting the potential for combinatorial strategies targeting PD-1, PD-L1, and RGMb to enhance immunotherapeutic outcomes. Finally, Sharpe closed by describing a CRISPR knockout screen in naive T cells used for adoptive therapy in tumor-bearing (or virally infected) mice to identify positive and negative regulators of antitumor/virus activity. In a screen with 899 gene knockouts, a number of expected targets were observed, but Stub1 emerged as a novel negative regulator. Stub1 is an E3 ubiquitin ligase that is known to be involved in regulating IFNγR and JAK1 in tumor cells, but had not been studied in T cells or other hematopoietic cells. Study of bone marrow chimeras to examine knockout of Stub1 in all hematopoietic cells showed that Stub1 KO in the immune system could prevent MC38 tumor growth. Also, proteomics of CD8+ T cells showed upregulation of the IFNγR, as observed in tumor cells, but also of the IL-27R. Further KOs in adoptively transferred CD8+ T cells showed that IL-27R upregulation was critical to the antitumor effect of Stub1 deletion, providing a mechanistic basis for promoting Stub1 as an attractive immunotherapy target.
Back to Top
Neoantigens and ERVs
Personalized cancer vaccines: Encouraging results and new opportunities - Catherine Wu, Dana-Farber Cancer Institute, Brookline, Massachusetts, USA
Cathy Wu’s talk was focused on how, among the wide range of immunotherapeutic strategies now available or in development as cancer therapeutics, vaccines are uniquely poised to address the key challenging attributes of tumor evolution and heterogeneity, as vaccines promote immunosurveillance and memory through supporting both existing and de novo responses. Key to the success of such vaccines is the identification of targetable neoantigens, which has improved with recent advancements in next-generation sequencing. Wu and colleagues initially focused on developing peptide-based neoantigen vaccines, which have proven to be safe, feasible, and immunogenic, showing particularly promising results in the adjuvant setting. To date, most clinical trials involving cancer vaccines have been limited to small numbers of patients, but large-scale, randomized, and treatment-controlled vaccine trials are currently in the works, especially with mRNA/LNP modalities, and will hopefully lead to clearer efficacy data. With these trials coming, Wu emphasized the importance of “bedside to bench and back again” research, in which patient outcomes inform continued research. Highlighting a recent phase I study, Wu described how patients with previously untreated renal cell carcinoma, a disease with a low tumor mutational burden, were treated with a personalized neoantigen vaccine (targeting up to 20 neoantigens) in combination with local ipilimumab. These patients showed no dose-limiting toxicities, and prolonged recurrence-free survival, with no disease recurrences at a median follow-up of just over 40 months. Extensive correlative research demonstrated robust immune responses, including responses against autologous tumors and responses specific for neoantigens derived from RCC driver mutations. Next, Wu highlighted two in-press trials, the first of which involved the treatment of 102 treatment-naive RCC patient with a combination of PD-1 blockade and vaccine, with the intent of evaluating response versus resistance. This study revealed that responding tumors showed increased expression of TLS signatures, while resistant tumors showed an emerging population of tissue-resident Slamf7+ exhausted CD8+ T cells; analysis of these two features together could be used to predict patient responses. The second new study that Wu described involved the search for new classes of targets, particularly those derived from transcription of endogenous retroviruses (ERVs), which are typically silenced, but can be re-activated in tumors. One such antigen that has already been identified is HERV-E (ERVE-4), which was initially recognized as a T cell target following allotransplant in patients with RCC, and is transcriptionally upregulated by HIF2. Following this path, Wu and colleagues demonstrated that HIF2 transcriptionally activated multiple ERVs, resulting in the production of immunogenic peptides. They also identified target-specific T cells in patients, and found that T cell responses to these peptides could be identified both in humanized mice and in patient samples from a prior study. Looking towards the future, Wu mentioned the possibility of using HIF stabilizers to induce ERV expression in tumors and drive neoantigen production. Finally, Wu described recent efforts in neoantigen identification by mass spectrometry in which data-independent acquisition (DIA) could be used to replace data-dependent acquisition (DDA) strategies, which are limited to the most abundant peptides. As DIA would require a user-defined peptide spectral library as a reference, Wu and colleagues recently developed such a library using a process called “Pepyrus”, which utilizes E. coli. These efforts proved to be successful and have been scaled up to 20,000 peptides, with Pepyrus-enabled DIA supporting the low-input detection of known neoantigen peptides. This strategy effectively identified patient-derived neoantigens in the melanoma setting, using generated patient sample-specific peptide libraries.
Immunogenicity and therapeutic targeting of recurrently mutated ‘public’ neoantigens - Christopher A. Klebanoff, Memorial Sloan Kettering Cancer Center, New York, New York, USA
Comparing the trade-offs between personal neoantigens – present in much greater numbers per patient, but requiring bespoke manufacturing, and likely not contributing to cancer cell fitness – and public neoantigens – derived from commonly found driver mutations that are widely found across patients and the tumor landscape, but fewer in number and subject to HLA restriction – Christopher Klebanoff turned his focus towards the latter. A pipeline was assembled utilizing (1) immunoproteomics to demonstrate antigen processing and presentation by prevalent HLAs, (2) next-generation targeted sequencing and patient sample acquisition via the BioTrust program, (3) immunogenicity testing and TCR retrieval from healthy donor PBMCs, and (4) eventual preparation of TCR-engineered T cells for patient treatment. The researchers focused on target mutations derived from both the highly prevalent KRAS point mutations at position G12 and NRAS mutations at Q61, as well as a gene fusion mutation in the serious, but not well served, pediatric/young adult disease, desmoplastic small round cell tumor (DSRCT). The DSRCT recurrent gene fusion between EWSR1 on chromosome 11 and WT1 on chromosome 22 created a novel junction peptide that could be detected by immunopeptidomics in the monkey COS-7 cell line, engineered to express the fused gene and HLA-A*03:01, HLA-A*03:02, or HLA-A*11:01. The junctional neoantigen was able to stabilize A2 cells expressing HLA-A*03:01 or HLA-A*11:01 and was readily detected by mass spectrometry in a tumor cell line (BOD) expressing HLA-A*03:01, at levels similar to those of HLA-A*03:01-binding peptides derived from the highly expressed filamin gene, and 4-fold higher than a comparable KRAS G12D-A*03:01 epitope. Two-color dextramer staining identified fusion peptide-specific T cells directly ex vivo in two patients, but these cells were highly exhausted (CD39+) and were not useful for TCR retrieval. Turning to in vitro screening with healthy donor-derived PBMCs and tetramer capture, a library of TCRs was isolated, and four TCRs were validated to bind to the fusion peptide. All the TCRs were of moderate affinity, and interestingly, one showed cross-reactivity between both HLA-A*03:01- and HLA-A*11:01-presented fusion peptides. The cross-reactivity was shown to be physiologically relevant in killing assays of isogenic cell lines expressing either HLA allele. Extensive amino acid substitutions in the peptide demonstrated shared anchor positions for both HLA allele types, and a common central core that sat above the binding groove and faced the TCR, with shared amino acids important for both TCRs. The TCR-engineered cell line specific for HLA-A*03:01 upregulated 4-1BB in response to a tumor explant from an HLA-A*A03:01 patient, and was cytotoxic ex vivo. In vivo, growth of the BOD cell line was effectively controlled by adoptive therapy with the TCR-engineered T cells, which was dependent on B2M expression in the tumor cells.
Towards personalized minimally-invasive T-cell therapies - Alena Gros, VHIO Vall D’Hebron Institute of Oncology, Barcelona, Spain
Following the recent accelerated FDA approval of Lifilieucel TIL therapy for unresectable or advanced melanoma, Alena Gros discussed what’s next for TIL therapy. One of the goals in this field is to develop more defined tumor-reactive cell products, and one strategy to accomplish this is to specifically expand neoantigen-reactive TILs. In a phase I clinical trial, Gros and colleagues expanded TILs, then selected for neoantigen-specific cells based on a coculture with B cells expressing predicted peptides. These neoantigen-specific TILs were then further expanded and used to treat patients. While small, this study has thus far shown promise, with evidence that TIL products indeed contained neoantigen-specific CD4+ TILs. A patient who was treated with only part of the full treatment protocol showed a partial response and stable liver metastases. Another strategy for developing tumor-targeted TIL products is to select neoantigen-reactive T cells through surrogate markers like PD-1 and CD39, which are typically co-expressed on neoantigen-reactive T cells. To do this in a way that was minimally invasive and would capture tumor heterogeneity, Gros and colleagues evaluated the use of circulating tumor DNA (ctDNA) compared to DNA from tumor biopsies for whole-exome sequencing and neoantigen identification. While most of the neoantigens identified between tumor biopsies and circulating tumor DNA were overlapping and clonal, some were unique to tumor biopsies, while others were unique to the ctDNA. Evaluating the small fraction of peripheral blood lymphocytes expressing PD-1 and CD39, Gros and team identified T cells recognizing neoantigens that were shared, as well as unique to either the tumor biopsy or ctDNA. When this system was applied across 8 patients with different cancer types, patients showed a wide range of neoantigen numbers across biopsies and ctDNA. Interestingly, looking at TILs versus peripheral blood lymphocytes, each expanded in IL-2, the researchers were able to identify substantially more neoantigen-reactive T cells and reduced exhaustion markers in peripheral blood T cells, suggested that circulating lymphocytes may have more capacity for expansion than their tumor-resident counterparts. Together, these results support the possibility of using ctDNA and lymphocytes derived from blood, rather than or in addition to tumor biopsies, to identify neoantigens and expand neoantigen-reactive T cell products for the development of better personalized cell therapies.
A new family of immunogenic antigens - Sebastian Amigorena, Institut Curie, Paris, France
The vast majority of the human genome does not code for specific genes, and about half of the “dark genome” consists of transposable elements (TEs). TEs are typically silenced in adult tissues, but can be de-repressed in cancer and can code for proteins. Investigating this, Sebastian Amigorena and colleagues evaluated 3164 cells from glioblastoma tumor cores and adjacent tissues across 4 patients. From this, they identified enriched TEs, and, following additional analysis and validation, identified 340 TE-encoded peptides. Comparison of tumor and healthy tissues revealed that a few of these peptides were either tumor-specific or were strongly enriched in tumors. Among these, the most reliable peptide was identified as MNO-P3, which was found to be overexpressed in 97% of glioblastomas in multiple scRNAseq datasets, and was highly specific to glioblastoma. Through screening, Amigorena’s team identified binders of the MNO-P3–MHC complex, and lead binders were further subjected to cross-reactivity analysis to confirm specificity. The lead binder was then incorporated into a bispecific T cell engager (BITE), which induced strong T cell-mediated killing of 3 different glioblastoma cell lines. Further, when the lead MNO-P3-targeting binder was incorporated into CARs, the resulting CAR T cells promoted rejection of orthotopic glioblastoma tumors in mice. In another example of peptides that arise from non-coding DNA, Amigorena highlighted non-canonical splicing events that occur in tumors, resulting in junctions between exons and transposable elements (JETs) and subsequently, production of novel tumor-specific peptides. Recently, Amigorena and colleagues have developed a pipeline to identify a wider variety of splicing events, including aberrant splicing events to other protein-coding regions and to regions in the “non-coding” genome. Compared to known public antigens PRAME and NY-ESO-1, MNO-P7 showed strong tumor expression and specificity. Another peptide, MNO-P12 (derived from the aberrant splice junction to a lncRNA involved in cancer progression and presented by HLA-A*03:01 and HLA-A*11:01), was also found to be highly expressed in multiple tumors and highly specific to tumor tissues. Moving forward, Amigorena hopes to identify binders for MNO-P12 and further evaluate it as a possible target for immunotherapy.
Immunogenicity of endogenous retroelements in cancer - George Kassiotis, The Francis Crick Institute, London, England
The repertoire of antigens in cancer ranges from common cancer-associated antigens, which are frequently shared, but are likely expressed at lower levels in healthy tissues, all the way to neoantigens, which are usually highly specific to an individual tumor in an individual patient. In the hunt for antigens that combine the best of both worlds, George Kassiotis has been investigating retrotransposable element (RTE)-derived antigens, which are commonly shared and do not require mutation, but are also highly specific to cancer, with no role in healthy tissues. In the human genome, RTEs are highly abundant and are scattered in and around genes. In cancer, aberrant transcriptional inclusion of RTEs can lead to expression of chimeric or truncated proteins that act as antigens. In an effort to identify these unique isoforms, Kassiotis and team assembled the cancer RTE transcriptome de novo, incorporating over a million transcripts. RTE overlapping transcripts identified this way were shown to be highly cancer-specific, with several transcripts shared across different cancer types. Analysis of the immunopeptidome validated the presence of RTE-derived antigens in melanoma, including multiple from a LincRNA, typically considered to be non-coding. RTE-derived peptides have also been identified by different mass spectrometry search tools. Investigating why cancers still develop despite the expression of antigens, Kassiotis and colleagues turned to T cell-intrinsic and -extrinsic factors. To study this, the researchers tested leukocytes from healthy donors against a panel of HLA-A2-restricted peptides, and used tetramer sorting and TCRseq to identify dominant TCRs. These were then transduced into Jurkat cells for testing of sensitivity and specificity to their target peptides. Unfortunately, most of the T cells exhibited affinities below the optimal range for effective target cell killing. Because the vast majority of transcription is tightly regulated for expression of exons, RTEs are expressed at very low levels relative to their abundance in the genome, due to epigenetic suppression. Repression of this epigenetic control can enforce increased expression of aberrant transcripts, but also of canonical retroviral proteins, which can assemble into high-order structures, including virulent particles that are far more immunogenic than individual proteins. Resurrection of viruses through recombination has been shown to induce leukemias in immunodeficient mouse models. Interestingly, a look at a wide range of tumor cell lines showed evidence of resurrected retroviral expression. In a highly immunogenic model of lung adenocarcinoma (KPAR), which induces TLSs and antitumor antibodies specific to a single Emv2 antigen from a viral envelope of eMLV – knockout of this virus made tumors more aggressive, while targeting the virus with antibodies was able to control tumor growth. Looking at retroviral elements that have recently been incorporated into the human genome, Kassiotis identified HER-K (HML-2), which retains enough of its structure to produce retroviral particles. HERV envelope proteins are also immunogenic, and have been found to be overexpressed in several human cancers.
Back to Top
Cytokines
Expanding the cytokine alphabet for immunotherapy - K. Christopher Garcia, Stanford University School of Medicine, Stanford, California, USA
Christopher Garcia’s talk focused on the cytokine-related aspects of the “toolkit” of available and novel protein reagents his lab is developing to enhance multiple immunotherapy approaches. In particular, Garcia discussed engineered cytokines and synthetic cytokine ligands and receptors, guided by the deep mechanistic insights into the intracellular signaling pathways. For example, cytokine assembly of receptor complexes triggers conformation changes, tyrosine kinase activation, and phosphorylation of various STAT molecules, which migrate to the nucleus as transcription factors. An early example of engineering of these systems was the modification of IL-2 to modulate STAT5 activation, which was shown to lead to more targeted activation of CD8+ T cells compared to other typically activated cells. A phase I study evaluated this modified cytokine as monotherapy in patients who had received prior immunotherapy, and showed safety, stimulation of antigen-activated CD8+ T cells, and encouraging clinical responses. Garcia then went on to present his attempts to expand the “alphabet” of cytokines beyond those that are naturally available, enhancing specificity and/or creating novel intracellular signaling opportunities. A first attempt was the creation of orthogonal cytokine/cytokine receptor pairs (“orthokines”) by modifying existing cytokines to exclusively pair with a modified receptor. An IL-2 orthokine successfully activated only T cells with the compatible modified receptor, allowing targeted activation of the T cells, and no off-target effects. A glimpse of the early clinical results with this molecule showed good activation of CAR19 T cells, with minimal activation of other IL-2 target cells (NK cells, Tregs), no inflammatory IL-6 induction, maintenance of a memory phenotype in the CAR T cells, and CAR infiltration into challenging high-volume CLL, setting the stage for future studies without lymphodepletion. The next generation of this approach was inspired by the observation that replacing the intracellular signaling domains (ICDs) of the IL-2 receptor with those of IL-9 led to the activation of 4 of the 5 STAT transcription factors. Stimulating such hybrid receptors with ortho-IL-2 was more effective than the standard ortho-IL2Rs. Moving to replacement of these ICDs with receptors not part of the common gamma chain family (such as domains from interferon receptors, the G-CSF receptor, or the EPO receptor) led to distinct STAT activation, potentially leading to novel biology. In mouse models, many of these hybrid receptors enhanced tumor control, and RNAseq of T cells from treated mice revealed clear phenotypes (Th2, memory) that were stimulated by these novel receptors. An intriguing example was a myeloid phenotype introduced into T cells with the hybrid G-CSF receptor, inducing “reprogrammed” phagocytic activity. The ortho-IL-22 receptor, which primarily signals through STAT3, promoted the expansion of stem-like T cells (TCF7, LEF1) and enhanced proliferation of these cells in lymph nodes and tumors. When the ortho-IL-22 receptor was engineered into NY-ESO-specific T cells, tumors were better controlled than the ortho-IL-2R. Finally, Garcia described TriKines – molecules with a cytokine fused to an antibody to bring another receptor to the signaling complex and expand the repertoire of activated STAT family members. In particular, an IL-21R Trikine enhanced STAT3 stimulation as desired (to enhance stem-like properties) and led to superior tumor control, without the toxicity of either IL-2 or IL-21, supporting further exploration of this broad potential combinatorial space.
Back to Top
CARs
Science, the Endless Frontier and CAR T cells - Steady gains and much potential - Crystal L. Mackall, Stanford University School of Medicine, Stanford, California, USA
Crystal Mackall discussed the progress and challenges of CAR T cell therapy, emphasizing its success in hematological malignancies while addressing the persistent hurdles in solid tumors. Recent advances in synthetic biology and cellular engineering are opening new possibilities for CAR T cell therapy, equipping researchers with tools to enhance T cell persistence, resist exhaustion, and optimize tumor targeting. Mackall highlighted several promising clinical trials demonstrating CAR T cell activity in solid tumors. GD2 CAR T cells have shown encouraging responses in neuroblastoma and midline gliomas, particularly in relapsed or refractory cases. IL-13Rα CAR T cells have exhibited activity in glioblastoma, while Claudin18.2 CAR T cells have shown promise in gastrointestinal cancers. Additionally, Claudin6 CAR T cells combined with a vaccine and GPC3 CAR T cells armed with IL-15 have demonstrated encouraging results in various solid tumors. Despite these advances, CAR T cell therapies must compete with alternative treatments such as antibody–drug conjugates (ADCs) and peptide–drug conjugates (PDCs), which are often cheaper and easier to deliver, meaning CAR T therapy must show significant clinical advantages to gain traction. To address these challenges, Mackall then introduced STASH-Select, a synthetic biology-based approach for higher-order cell engineering that allows precise selection of engineered immune cells, requiring expression of more than one introduced CAR or other payload. This system enabled researchers to generate CAR T cell products with high purity and yield while maintaining compatibility with GMP manufacturing. STASH-Select requires transducing at least two vectors, each with one of two CARs/payloads and also containing a P2A sequence followed by a second “selecting” protein. One of these proteins is an endoplasmic reticulum (ER)-retention tag, which retains the surface construct in the ER. The selecting protein on the other vector encodes a protease that cleaves the ER retention tag, allowing the translocation of the ER-retained protein to the cell surface, and enabling the isolation of properly dual-payload-engineered cells. Alternatively, a resistance gene-based system can be used, where resistance is only conferred in the presence of a protease. A three-way AND gate system can distribute the protease across two vectors, allowing the creation of tri-cistronic vectors that enable the development of “Super CAR T cells” capable of expressing multiple tumor targets, cytokines, and a suicide switch for enhanced safety. Mackall discussed the application of STASH-Select in medulloblastoma, a prototypic embryonal brain tumor, where local delivery of CAR T products targeting three specific proteins (B7H3, GD2, and GPC2) appeared to provide the most comprehensive coverage across tumor and patients. However, tri-specific CAR T cells showed reduced potency and were prone to exhaustion, requiring further optimization. To enhance function, c-Jun overexpression was introduced, as it has been shown to improve CAR T cell resistance to exhaustion. Although c-Jun alone was only modestly successful, when combined with MED12 knockdown, tri-specific CAR T cells demonstrated improved persistence and functionality. Additional potency-enhancing modifications will be necessary to ensure the fitness of these CAR T cells. Mackall concluded by emphasizing the importance of integrating these enhancements strategically to develop next-generation CAR T cell therapies capable of overcoming the complex challenges posed by solid tumors.
Direct in vivo transduction of T and NK cells to treat B cell malignancies - Saar Gill, University of Pennsylvania, Philadelphia, Pennsylvania, USA
In recent years, CAR T cells have emerged as powerful immunotherapies, particularly against B cell malignancies. However, Saar Gill noted that the time and cost of manufacturing CAR T cells are both high, which limits their clinical use. To address this, researchers have evaluated CAR T cell products, and have found that the age and quality of cells might be more important than the quantity. Notably, younger T cells manufactured over a shorter duration have shown efficacy at lower doses than those manufactured for longer. Taking this concept further, Gill and colleagues wondered whether CAR T cells could be manufactured in vivo through gene therapy. They noted that transduction is more efficient at biological temperature, and that while T cell activation is typically necessary for transduction, there are alternative protocols that enable successful transduction of non-activated T cells, resulting in T cells with enhanced antitumor performance at lower doses. Considering whether injecting lentiviruses directly into patients would be safe, Gill noted evidence from HIV showing that lentiviral vector binding to cells can be highly cell type-specific, and that lentiviral integration into the genome is generally safe, and in some cases, even beneficial, though, like with ex vivo transduction, may cause rare cases of leukemia development. Putting the possibility of generating CAR T cells in vivo to the test, Gill and team developed a lentiviral vector that was redirected from LDL to CD7, which is expressed on T and NK cells. Encoded in the vector was a transgene for a fully human CAR targeting CD20. In humanized NSG mice engrafted with human B and T cells, treatment with the vector led to expression of the CAR in T cells in the blood, spleen, bone marrow, and liver; expression of the CAR in NK cells, which were present at low levels; and B cell depletion. In a lymphoma model, lentiviral treatment showed antitumor efficacy and eradication of lymphoma. In monkeys, treatment led to B cell aplasia, and to CAR+ T and NK cells in spleens of treated animals. Based on this study, Gill and colleagues initiated a phase 1 clinical trial in Australia for patients with B cell malignancies that had relapsed after at least 2 prior therapies, which could have included a prior CAR T cell treatment. To date, 3 patients have received treatment – 2 at dose level 1 and 1 and dose level 2. Thus far, no instances of CRS, neurotoxicity, or infusion reactions have occurred. In patients treated at the lower dose, B cell counts did not change, but in the patient who was treated at the higher dose (and who had previously relapsed after treatment with anti-CD19 CAR T cells), B cell aplasia was observed, suggesting very early, but promising results.
Back to Top
T cell Responses
Immune evasion – from CD8 to CD4 T cells - Annette Paschen, University Hospital Essen, Essen, Germany
Annette Paschen focused on the dynamic interplay between tumor cells and the immune system, and on a stepwise view of how melanoma metastases can evolve to escape both CD8+ and CD4+ T cell-mediated recognition. Beginning with a patient harboring multiple metastases, Paschen described how an initially immunogenic lesion (86a) displayed robust HLA-I expression and had responded to an IFNα, IL-2 peptide vaccine. However, multiple other lesions (86b, c and f) emerged over time, and were resistant to immunotherapy. Upon generation of cell lines from these metastatic lesions, the responding lesion 86a showed higher expression of MHC-I molecules, while resistant lesions 86b and 86f lacked MHC-I due to B2M losses, which were distinct in each lesion. 86c did express MHC-I, suggesting an alternative mechanism of immune escape. Four distinct CD8+ T cell-recognized neoantigens were identified using 86a as target, but these T cells failed to recognize 86c. Further genetic analysis revealed HLA haplotype loss in the 86c lesion prevented neoantigen presentation on the relevant HLAs while maintaining the non-relevant HLA. T cell-mediated killing could be restored by reintroducing the lost HLA alleles. Importantly, TILs were able to recognize the original neoantigens, indicating that the loss was specific to 86c, and not a general failure of the immune system. Shifting focus to CD4+ T cells, Paschen observed constitutive MHC-II expression in 86a, while 86c expressed MHC-II only upon IFNγ treatment. Autologous CD4+ TILs were robustly activated directly by 86a, and activation was blocked by antibodies against HLA-II, confirming the specificity of the response. However, 86c, despite attempts to induce MHC-II expression with IFNγ, failed to elicit CD4+ T cell responses. Further investigation revealed not only the loss of MHC-I, but a deletion encompassing the MHC-II region in 86c. Activation of CD4+ TILs could be successfully restored by transfecting 86c with MHC-II genes. The researchers also observed heterogeneity in constitutive MHC-II expression in another patient (Mel-61), who also had multiple metastases, and observed that this heterogeneity correlated with expression of the CIITA gene – a known MHC-II regulator. HLA-II expression was sufficient for autologous CD4+ TIL-mediated killing, which could be blocked by MHC-II inhibition. Further investigating the impact of IFNγ on MHC-II expression, the researchers found that IFNγ treatment upregulated MHC-II in 4 cell lines, but failed to do so in Mel61g, demonstrating the intrapatient heterogeneity in the IFNγ response. Mel-61g showed a defect in the JAK-STAT pathway due to an acquired mutation in JAK1. Wild-type JAK1 transfected into the Mel-61g cell line restored MHC-II expression with IFNγ, and restored the CD4+ T cell response. This was confirmed in another patient with stage VI cutaneous melanoma (Mel-36), which contained a mixture of JAK1-wild-type and JAK1-mutant clones. The JAK1-mutant cells did not upregulate MHC-II in response to IFNγ, while the JAK1-wild-type cells showed constitutive MHC-II expression, even in the absence of IFNγ. To test whether JAK signaling also contributed to the constitutive expression of MHC-II, the researchers treated JAK1-WT cells with Ruxolitinib (a JAK inhibitor) and saw downregulation of JAK pathway signaling and MHC-II expression, suggesting tumor cell-intrinsic JAK signaling is essential for MHC-II expression on melanoma cells, independent of IFN signaling. The researchers further asked whether B2M loss variants could express constitutive HLA-II in another patient with melanoma (Mel-21). Co-culture experiments with autologous CD4+ PBLs showed activation of CD4+ T cells in the B2M-loss variant, and this recognition was blocked by an anti-MHC-II antibody. Paschen further demonstrated that the constitutive HLA-II phenotype dominates in a subgroup of melanoma metastases, and, with some caveats and further work, CD4+ T cell-mediated constitutive HLA-II targeting may be a viable alternative in these tumors.
Translational control of the TME cells - therapeutic opportunities - Caroline Robert, Gustave Roussy, Paris, France
With melanoma, it is not uncommon to see tumors that are half-disappearing, where the immune system spontaneously destroyed parts of a tumor, but ultimately failed to control it. Treatment with immunotherapies have been particularly effective in melanoma, sometimes aided by second line BRAF- and MEK-targeted therapies, though relapse and resistance can still occur through various mechanisms. In her talk, Caroline Robert highlighted RNA translation as a mechanism of resistance that may be underestimated. For example, studies have shown that the initiation of RNA translation, mediated by binding of elF4E to the CAP, is the rate-limiting step in the translational control of mRNA, which allows cells to quickly adapt to various stressors. Overexpression of elF4E can induce spontaneous lymphomagenesis, while suppression of elF4E can reduce tumor growth. Robert and colleagues have also identified increased elF4F complex formation as a mechanism of resistance in the context of both targeted therapies and immunotherapies. To better understand how cells remain invisible under treatment, and how this might play a role in late relapses, Robert evaluated in vitro and in vivo models in which tumor cells develop drug resistance. Interestingly, “persister cells” showed low translation overall, but a small subset of genes were highly translated, with each showing contributions to cellular resistance. To track persister cells in patients, the researchers utilized antibodies that target the proteins encoded by the upregulated genes that were identified for their involvement in resistance. When these genes were expressed early after treatment, patients were more likely to have poor response and survival outcomes. Coculture experiments with immune cells and persister cells revealed that the supernatant contained a factor that reduced the activation and tumor-killing capacity of lymphocytes, instead enhancing markers of exhaustion and dysfunction. Analysis of cytokines in the melanoma secretome after just two weeks of targeted therapy revealed many changes, including strong upregulation of macrophage migration inhibitory factor (MIF) in the supernatants of persister cells compared to parental tumor cells. Cancer cell expression of MIF is a strong negative regulator of innate immunity and tumor control. However, MIF mRNA, translation, and stability were not increased in tumor cells. Instead, Robert and colleagues found that persister cells upregulated mRNA expression of ABCA7 – a transporter that enables the export of MIF and the subsequent recruitment of suppressive macrophages. However, use of an elF4A inhibitor decreased ABCA7 expression, which reduced MIF secretion. In a mouse model of persistence, elF4A inhibition improved antitumor responses to targeted therapy. Further, efF4A was found to control expression of T cell activation and exhaustion markers, and inhibition of efF4A improved the quality of T cells and their capacity for melanoma cell killing. For Tox, the impact on translation was traced to its long and highly structured 5’ UTR. Looking in patients who were responders and non-responders to PD-1, Robert showed that ex vivo treatment of a live melanoma section with anti-PD-1 plus an elF4A inhibitor, decreased viable tumor cells and increased tumor necrosis. In mice, this same combination increased T cell infiltration and antitumor efficacy.
Impact of tertiary lymphoid structures on the tumor-specific T cell landscape - Daniela Thommen, Netherlands Cancer Institute, Amsterdam, Netherlands
Daniela Thommen emphasized the crucial role of the tumor microenvironment (TME) in regulating T cell functions and responses to immunotherapy. To investigate these mechanisms, Thommen and team developed the Patient-Derived Tumor Fragment (PDTF) platform – an ex vivo model that cultures fragments of a tumor sample in individual wells, preserving the native tumor architecture and immune composition. Using PDTFs, the researchers assessed immune activation following PD-1 blockade across multiple cancer types. To account for intratumoral heterogeneity, multiple fragments were cultured and pooled for analysis. The anti-PD-1-treated PDTFs revealed two distinct groups of tumors: responders, which exhibited increased expression of activation markers and cytokines, and non-responders. PDTF responses correlated with patient clinical outcomes, highlighting the model's predictive potential. Further analysis of individual tumor fragments revealed spatial heterogeneity in immune responses, with some fragments from the same patient showing activation, while others remained unresponsive, despite comparable T cell infiltration. In lung cancer, intratumoral T cells reside in distinct neighborhoods, particularly in tertiary lymphoid structures (TLSs). Drawing a comparison to the Netherlands’ population density in cities and in the countryside, Thommen highlighted the spatial distribution of T cells in higher clusters, as in TLSs, and dispersed at lower density within tumors. To better understand the role of location and tissue context on intratumoral T cells, the team performed TCR repertoire sequencing and single-cell transcriptomics on microdissected regions, comparing T cells from TLSs and tumor regions. Highly expanded T cell clones were present in both compartments, suggesting that spatial variation in immune responses was not driven by distinct TCR distributions. Instead, scRNAseq revealed that T cells in TLSs were enriched for progenitor-like states, whereas tumor-resident T cells exhibited more terminally differentiated and dysfunctional phenotypes. The most expanded shared clones were enriched for the dysfunctional states, and individual clonotypes were able to exist in different cell states. To further assess the distribution of the cell states, the researchers leveraged six publicly available spatial transcriptomic datasets, and found that TLSs supported a reservoir of progenitor-like T cells in tumors, while dysfunctional states were present outside of the TLSs. To investigate how TLS and non-TLS CD8+ T cells respond to PD-1 blockade, the researchers integrated gene expression signatures to infer TLS presence in tumor fragments, and assessed their response to PD-1 blockade. A TLS gene expression signature discriminated TLS and non-TLS regions in spatial transcriptomic data and micro-dissected tumor areas. While TLS-rich PDTFs did not show increased baseline cytokine production, they exhibited enhanced activation upon treatment, particularly in terminally exhausted T cells, suggesting that TLSs contribute to immune reactivation by maintaining a pool of T cells that can be reinvigorated by checkpoint blockade. Further investigations into dysfunctional T cells revealed that, despite high expression of effector genes, these cells exhibited a post-transcriptional barrier, limiting protein translation. PD-1 blockade appeared to overcome this barrier, restoring IFNγ secretion and enhancing T cell functionality. Overall, the findings highlighted the spatial organization of immune responses in tumors, with TLSs playing critical roles in supporting both progenitor-like and dysfunctional T cell populations.
T cell dysfunction - Andrea Schietinger, Memorial Sloan Kettering Cancer Center, New York, New York, USA
Andrea Schietinger challenged the conventional view of T cell dysfunction, advocating for a more nuanced perspective that differentiates it from mere T cell exhaustion. Aiming to prevent T cell dysfunction, Schietinger and team knocked out TOX in T cells and found that TOX KO T cells did not establish transcriptional and epigenetic programs of exhaustion. However, despite lacking the typical exhaustion markers, these cells were functionally impaired. While initially displaying robust effector functions, TOX-deficient T cells were prone to rapid attrition and activation-induced cell death. This finding highlights the critical role of TOX-driven exhaustion in maintaining T cell persistence within the tumor microenvironment (TME). Schietinger emphasized that "TOX" is not simply a driver of exhaustion, but a key factor for T cell survival during chronic TCR stimulation. The expression of inhibitory receptors, often considered a hallmark of exhaustion, can also be interpreted as a protective mechanism. These receptors act as negative feedback regulators, preventing excessive activation, and allowing cells to persist in a state of controlled dysfunction, rather than succumbing to complete functional collapse or cell death. Schietinger proposed two modules downstream of TCR: a TOX-driven exhaustion phenotype that allows T cells to persist in tumors, and a TOX-independent phenotypic dysfunction that is associated with loss of effector function. In addition to her work on T cell exhaustion, Schietinger’s team has explored strategies to optimize T cell subsets for immunotherapy. Drawing inspiration from autoimmune diseases, where certain T cell subsets are pathogenic, Scheitinger noted that the genetic allele associated with the disease risk is MHC-II, implicating CD4+ T cell involvement. To examine the role of CD4+ T cells in enhancing CD8+ T cell-mediated tumor cytotoxicity, the researchers engineered cancer cells to express both CD8+ and CD4+ tumor antigens. While adoptive transfer of CD8+ T cells alone failed to eliminate tumors, co-transfer of CD8+ T cells and in vitro-activated CD4+ T cells resulted in tumor eradication across several mouse models. Mechanistically, CD4+ T cells did not increase the number of CD8+ T cells, but rather, enhanced their functionality. Crucially, this suggested that the spatial organization of CD4+ and CD8+ T cells within the tumor was required for this functional enhancement and tumor elimination. Depletion of CD11c+ APCs within the tumor resulted in tumor escape from CD8+/CD4+ T cell-mediated cytotoxicity. The researchers also investigated whether the formation of CD8-CD4-APC triads were necessary for CD8+ T cell reprogramming. Fluorescent labeling revealed that triads were present more frequently in responding versus non-responding tumors, suggesting that triad formation is critical for CD8+ T cell cytotoxicity. Additionally, experiments showed that a mixture of APCs with either MHC-I or MHC-II expression alone was insufficient for effective antitumor immunity, further reinforcing the necessity of CD4+ and CD8+ T cells engaging with the same APC to mount a durable immune response. CD4+ T cell depletion at later stages abolished antitumor immunity, emphasizing the need for sustained interactions between CD4+ and CD8+ T cells and their APCs. Clinical data also supported these findings. In patients with malignant pleural mesothelioma treated with immune checkpoint blockade (ICB), the presence of intratumoral triads was observed in responding patients, with CD8+ T cells in these triads exhibiting an activated and cytotoxic phenotype. Schietinger then presented her lab's work investigating the role of TOX in CD4+ T cells. They found that TOX is expressed in activated CD4+ T cells and plays a pivotal role in Th1 polarization. Overexpression of TOX in Th0 cells drove their differentiation into Th1 cells, increasing IFNγ production, without affecting TNFα levels. Further transcriptional and epigenetic analyses confirmed that TOX is a master regulator of Th1 differentiation and IFNγ production. Reanalysis of published data revealed that CXCL13+ CD4+ T cells, which express high levels of TOX and IFNγ, were enriched in patients with bladder and hepatocellular carcinoma that responded to ICB. Schietinger concluded by proposing that TOX serves as a marker for a subset of IFNγ-producing CD4+ T cells, which could act as a positive prognostic indicator of ICB responsiveness in human tumors.
Back to Top
Tumor Immune Microenvironment
Spatio-temporal regulation of tumor associated macrophages - Florent Ginhoux, Gustave Roussy, Villejuif, France
Macrophages are the major immune cell type in most cancers, but are often seen as the “bad guys” in immune oncology, Florent Ginhoux argues that their role is far more complex than that, with roles in matrix remodeling, immunomodulation, EMT, angiogenesis, and intravasation. These roles often vary depending on the macrophage’s origin, tissue environment, the presence of inflammation, and the duration of exposure to particular environmental factors, which can contribute to a range of phenotypes and activities that go well beyond the paradigm of M1 and M2. Ginhoux noted that macrophages can either be of embryonic or adult origin. To better track macrophages in mice, including where they go and how they contribute, Ginhoux and colleagues utilized a monocyte fate mapper model for time stamping experiments. By tracking common monocyte progenitors (cMoPs) through tagging of Ms4a3, this model revealed that in several tumor models, TAMs were mostly monocyte-derived, but the resident tissue macrophages (RTMs; express CD206) in adjacent tissues were of embryonic origin, and were spatially localized at the tumor periphery. Similar spatial segregation was observed with immunostaining of human PDAC. Looking at heterogeneity within monocyte-derived TAMs, Ginhoux and colleagues used their time stamping model and identified three distinct clusters of TAMs – TAM1, TAM2, and TAM3 (none of which aligned with M1 or M2 phenotypes). Predicting the relationships between them showed that TAM1 is likely an intermediate phenotype, which diverges into either TAM2 or TAM3, representing lineage branching. This was validated using Infinity Flow (a machine learning enhanced flow cytometry approach) with 270 markers to determine the time relationship between TAMs, and using CODEX to map TAM locations. Interestingly, this also showed that the different subpopulations of TAMs occupied different tumor niches, with hypoxic macrophages localizing to the necrotic tumor core. Similar results were observed in human PDAC. Overall, these results broaden our understanding of macrophages both within and outside of tumors, and their complexity needs to be embraced in order to properly inhibit their immunosuppressive functionalities, and/or support their antitumor potential in cancer.
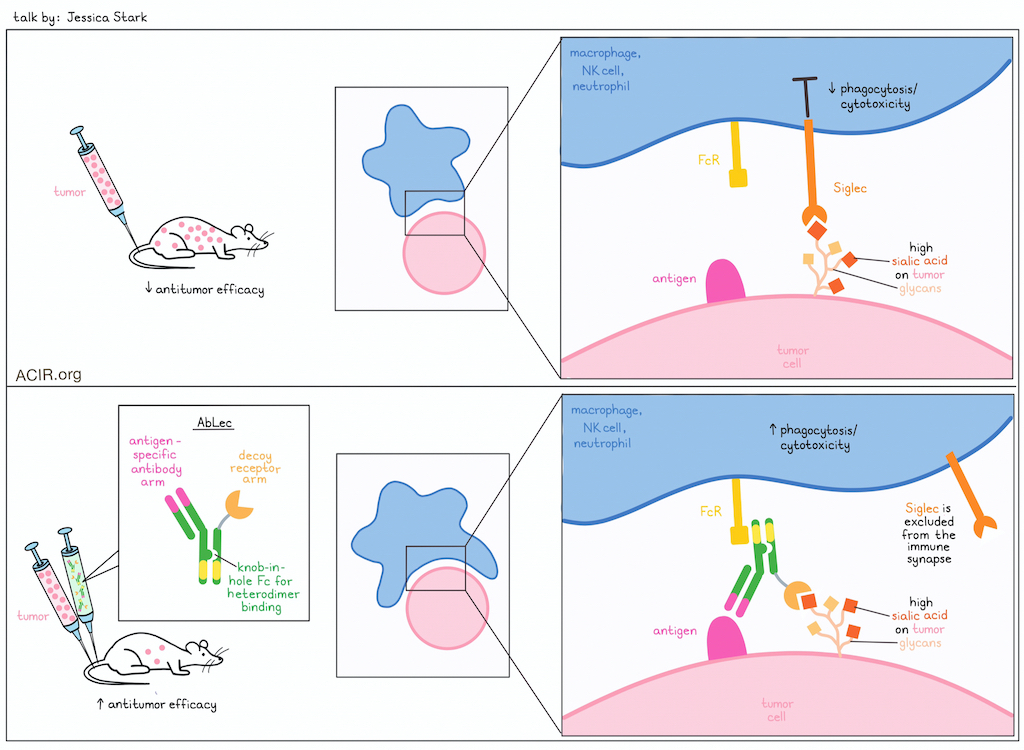
Antibody lectin chimeras for glyco-immune checkpoint blockade - Jessica Stark, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA
While checkpoint blockade is currently used in roughly half of patients with cancer, tumors often resist treatment or evade detection, limiting responses. Investigating mechanisms of immune evasion, Jessica Stark highlighted the importance of cell surface glycans in immune recognition, and aberrant glycosylation in tumors. In particular, tumors frequently and substantially upregulate the monosaccharide sialic acid, which is recognized by receptors known as Siglecs on multiple immune cell subsets. Like PD-1, Siglecs send signals that dampen immunity when they encounter their ligands, and are associated with worse clinical outcomes, suggesting that upregulation of sialic acid on tumor cells is likely a mechanism of immune evasion. While Siglecs are promising target for immunotherapy, attempts at targeting them have been limited, as antibody targeting has a high affinity, but limited glycan binding, and decoy receptor molecules have strong glycan binding, but are limited by low affinity. In an effort to get the best of both strategies, Stark and colleagues developed AbLec molecules, which are heterodimeric bifunctional molecules (based on Fc-engineered knob-in-hole technology) with a high-affinity HER2-targeting antibody arm and a decoy receptor arm targeting either Siglec-7 or -9. These AbLecs bound to human cancer cell lines at levels on par with the parent antibody, and effectively blocked Siglec binding in human cancer cell lines. Further, the researchers found that AbLecs potentiated cytotoxicity in NK cells and phagocytosis in macrophages and neutrophils, and was partially dependent on FcγR binding. In a Siglic-7/9 and FcγR humanized mouse model of lung metastasis, AbLecs reduced metastatic burdens and dampened Siglec-mediated immunosuppression. A closer look at the immune synapse revealed that the presence of Siglec-7 in the early moments of the encounter inhibited phagocytosis, and that the use of AbLecs excluded Siglec-7 from the immune synapse, allowing phagocytosis of the target cells to proceed. Finally, Stark emphasized the generalizability of this platform, as different antibodies and decoy receptors could be interchanged depending on the cancer setting, allowing for a flexible platform to further develop Siglec-targeting, or even dual-checkpoint-targeting immunotherapies.
In Situ Approaches
Intratumoral immunotherapy mimicking viral infections - Ignacio Melero, Clínica Universidad de Navarra, Pamplona, Spain
Inspired by the effectiveness of immune responses during most viral infections, Ignacio Melero focused on how T cell priming can be effectively accomplished by turning the tumor bed into a vaccine site with Human Intra-tumoral Immunotherapy (HIT-IT). A key objective is to act locally, where there is a high abundance of tumor antigens and limited opportunity for toxicity, but also to have global effects to control distant micro-metastatic disease. Historically, this approach has its roots in the work of William Coley, and there are two FDA-approved therapies for local disease (intravesicular BCG for superficial bladder cancer and imiquimod for basal cell carcinoma of the skin). Inspired by these successes, in the late 2010s, there began a significant clinical effort with multiple stimulatory modalities (oncolytic viruses, TLR agonists, checkpoint inhibitors, etc.), but no new registrations resulted. Four newer modalities are showing progress though. First is intratumoral injection of an immunostimulatory RNA (polyI:C) – mimicking an RNA virus in the tumor – delivered in a PEI-based nanoparticle (BO-112). Strong antitumor activity was shown in BO-112-injected tumors in multiple animal models, although distal effects were more limited. Correlative analysis of draining lymph nodes showed improved T cell infiltration, an increased CD8/Treg ratio, and a typical effector T cell phenotype. A phase I trial of BO-112 and anti-PD-1 therapy in patients anti-PD-1-refractory melanoma and NSCLC showed responses. A subsequent phase 2 trial, demonstrated further responses, including complete responses (CRs) in approximately 25% of patients, and regression of both injected and distal lesions. Combining local radiotherapy with BO-112 and systemic anti-PD-1 in animal models also showed good efficacy locally and distally. It was also noted that untreated tumors became sensitized to radiotherapy, which inspired the ongoing BOLT trial. Melero then described studies with the immunogenic cell death inducer Tiliganol tiglate, a Protein Kinase C agonist isolated from the seeds of a fruit in the Australian rainforest. Direct intratumoral injection of this molecule in murine tumors resulted in local and distal effects, including immune memory. Studies in companion dogs with mast cell sarcoma were very effective, with over 75% complete responses (CRs); this is now an approved veterinary therapy in AU, the EU, and the US. In clinical studies in 26 patients with subcutaneous sarcoma, 10 CRs and 8 PRs were observed. In a subsequent ongoing phase II trial, there is evidence of effects on distal lesions in a leiomyosarcoma. Turning to intratumoral cytokines, Melero described a rationale for the use IL-12, which is under development using multiple delivery modalities, and also for IL-18, particularly a “decoy-resistant” IL-18 (IL-18DR) that is altered to prevent binding to the inhibitory IL-18 binding protein, which is highly expressed in tumors. Animal studies demonstrated good local and distal tumor control ( dependent on CD8+ T cells and Batf3+ DCs), induced significant remodeling of the TME, and improved with anti-PD-1 therapy. Finally, Melero described some newer work using TCR- or CAR-T cells to co-deliver various cytokines/stimulators (IL-12, IL-18DR, 41BBL) directly to tumors, with exciting animal data.
STINGing tumors to improve cancer immunotherapy - Zhijian James Chen, UT Southwestern Medical Center, Dallas, Texas, USA
Zhijian James Chen introduced the intricate workings of the cGAS-STING pathway, a critical component of the innate immune system that detects cytosolic DNA and triggers a robust innate immune response. Upon binding to foreign or misplaced double-stranded DNA (dsDNA) in the cytosol, cGAS undergoes a conformational change, leading to the synthesis of the second messenger, cyclic GMP-AMP (cGAMP), from ATP and GTP. This small molecule serves as a critical signaling intermediate that activates STING, a transmembrane protein located in the endoplasmic reticulum. Activated STING subsequently recruits and activates TBK1, which phosphorylates IRF3. Phosphorylated IRF3 then translocates to the nucleus, where it induces the transcription of type I IFNs and other inflammatory cytokines. Chen highlighted that cGAS activation is sequence-independent, and instead relies on recognition of the sugar–phosphate backbone of DNA, enabling cGAS to detect a wide range of pathogenic DNA, including that from viruses, bacteria, and tumors. However, this broad recognition also poses a risk for aberrant activation in autoimmune diseases. Looking at the role of STING activation in cancer immunotherapy, Chen highlighted preclinical studies in lung cancer mouse models in which systemic administration of cGAMP promoted robust CD8+ T cell responses, leading to tumor regression in anti-PD-L1-resistant tumors. However, early clinical trials of STING agonists have yielded disappointing results, largely due to poor systemic bioavailability, as these trials were limited to intratumoral injection, which fails to generate an abscopal effect. To address these challenges, Chen and his group used an antibody–drug conjugate (ADC)-based approach to systemically deliver and selectively target STING agonists to tumors. By conjugating a modified STING agonist (IMSA172) to a tumor-targeting antibody (anti-EGFR), the team developed the ɑEGFR-172 ADC, which achieved localized drug release within the tumor microenvironment, enhanced efficacy, and reduced systemic toxicity. This anti-EGFR/STING ADC potently induced interferon responses and synergized with an anti-PD-L1 antibody, further enhancing antitumor responses and leading to prolonged survival in a B16-EGFR tumor model. Mechanistically, the STING ADC activated CD4+ and CD8+ T cells in the tumor, along with dendritic cells, which migrated to tumor-draining lymph nodes; it also polarized macrophages from M2 to M1. Chen then introduced a lipid nanoparticle (LNP)-based delivery system for STING agonists, designed to improve drug stability and optimize immune activation. Based on a mutated form of cGAS (R241E) that reduces interaction with histones, a constitutively active cGAS (R241E) mRNA LNP facilitated targeted release in tumors, ensuring sustained immune stimulation. To further enhance efficacy, the researchers combined the cGAS (R241E) LNP with cytokines, including IL-12. Membrane-tethered IL-12 constructs allowed localized cytokine release in tumors, avoiding systemic toxicity while maintaining therapeutic efficacy. Preclinical studies demonstrated that this combination therapy induced potent T cell infiltration into tumors, enhanced IFNγ production, and exerted antitumor effects in both treated and untreated tumors in a CD8+ T cell-dependent manner. PD-L1 blockade further enhanced the antitumor effects of cGAS/IL-12 mRNA LNPs and promoted long-term survival in the B16 mouse model.
Back to Top
By Lauren Hitchings, Shishir Pant, and Ed Fritsch



