
Last week, the ACIR team attended the Koch Institute Immune Engineering Symposium in Cambridge, Massachusetts. This week’s extensive special feature covers select talks from the conference.
Robert Schreiber opened the symposium by examining the role of MHC II and CD4+ T cells in tumor control, particularly with the backdrop that many tumors do not express MHC II. Schreiber’s team applied their newly developed MHC II prediction algorithm to their immune-edited T3 sarcoma mouse model, in which the role of MHC I dominant and subdominant neoantigens has been well established, to identify multiple MHC II neoantigen candidates and search for CD4+ T cells in mice bearing freshly implanted tumors. Only one strong response arose targeting a mutated integrin (mItgb2; N711Y) and subsequent studies validated mutant specificity. Turning then to gene-edited tumor studies with either or both of mItgb2 and mLama4 (a strong mutated MHC I epitope) inserted into the oncogene-driven Kras/p53 cre-inducible (KP) tumor line, which is poorly immunogenic and does not respond to checkpoint blockade nor generate immune memory, Schreiber was able to show with irradiated target cells as antigens that KP cells expressing either MHC I or II epitope alone were poorly protective for subsequent tumor implant, but that both neoantigens prevented tumor growth in most mice. Confirmatory studies demonstrated strong enhancements in the generation of CTLs targeting mLama4 only in the presence of the mItgb2 CD4+ T cells, indicating a strong role for effective CD4+ epitopes in CTL priming and/or maturation, and suggesting that the balance of effective CD8- and CD4-targeting neoantigens may be critical to tumor control.
Tyler Jacks used his Kras/p53 cre-inducible (KP) tumor model to begin studying the role of tumor-extrinsic factors (immune cells, stroma, angiogenesis, TME) in tumor control following insertion of two model neoantigens (OVA and the SIY epitope). Although these strong epitopes slowed tumor growth early after oncogene induction, by 16 weeks that tumor growth control was lost despite continued antigen expression by the tumor, which could be traced to a significant increase in regulatory T cells. In collaboration with Aviv Regev, Jacks used single-cell RNAseq to analyze the phenotype of CD8+ T cells over time. t-SNE plots showed that the antigen strength (OVA > SIY) affected phenotype and that the phenotype evolved over time toward a more dysfunctional state (PD-1+, LAG3+, Ki67-), which correlated with known markers of dysfunction in human melanoma and with increasing unresponsiveness to checkpoint therapy. Within these dysfunctional T cells, the transcription factor EGR2 was induced, and adoptive T cell transfer experiments with EGR2 knockouts showed enhanced proliferation/maintenance, reduced inhibitory co-receptor induction in tumor-bearing mice, and a distinct transcription profile compared to EGR2+ T cells. A similar CRISPR knockout screen of other immune-relevant genes in this system revealed more genes which negatively (TGFβR, Arid1a, PD-1) or positively (IL2Rγ) affected tumor growth or the T cell exhaustion phenotype. Interestingly, other than PD-1, no other single co-inhibitory receptor knockout had an impact, but dual knockout of LAG3 and TIM3 was effective.
In the case of poorly immunogenic tumors with few or no known neoantigens, shared, overexpressed, tumor-self antigens may be the only targetable option. Such antigens are often recognized by T cells with low affinity. Nevertheless, Stephanie Dougan demonstrated in tumor-bearing mice that high- and low-affinity T cells were equivalent in their ability to control tumors. The priming and function of low-affinity T cells could be further augmented by several methods, including the combination of radiation and CD40 agonist, or the inhibition of IAP (inhibitor of apoptosis protein), which mimics the effect of TNF family costimulatory receptor signaling. While IAP antagonism enhances the function of many immune cells, it had no effect on pancreatic tumors in vitro. However, in vivo, IAP antagonists cured mice of pancreatic cancer and established immunological memory. In fact, IAP antagonists outperformed checkpoint blockade in T cell-cold tumors, and the combination of IAP antagonists with checkpoint blockade was curative in T cell-high tumors. To understand the difference between the in vitro and in vivo results, Dougan dove into mechanistic studies. Her team found that while CD8+ T cells were required for antitumor response, surprisingly, neither MHC class I expression nor IFNγ sensing by pancreatic tumor cells were necessary. Instead, intratumoral macrophages were critical for the in vivo response in pancreatic tumors. While IAP antagonism did not directly affect macrophage phagocytosis in vitro, it increased phagocytosis in vivo. Dougan closed with a model based on her work in which only a small number of properly activated antigen-specific T cells (through non-canonical NF-κB signaling in DCs) could prime innate immune cells to control tumor independently of dramatic T cell expansion or MHC I expression on tumors.
Max Krummel updated the audience on the importance and differential roles of the myeloid compartment, particularly the key antigen-presenting cells, in affecting T cell numbers and phenotype, as shown in a video of T cells swarming around cDC1 dendritic cells, but aimlessly stuck around tumor-associated macrophages in the tumor margins. Analysis of clinical tumor samples by flow cytometry or FACS followed by RNAseq revealed important populations contributing to CD8+ and CD4+ T cell responses. For CD8+ T cells, response to anti-PD-1 therapy was strongly tied to presence of the cDC1 (BDCA3+, CD103+, Batf3-dependent, IL-12-producing) population. BDCA3+ DC numbers correlated with the level of intratumoral FLT3L, which was produced by NK cells, and with improved tumor control. For CD4+ T cells, the presence of cDC2 (irf4-dependent, BCDA1+) DCs was critical. cDC2 cells must migrate to lymph nodes for CD4+ T cell induction and the number and phenotype of these cells were strongly controlled by regulatory T cells. Interestingly, in a tumor type such as melanoma with a low Treg/CD3+ intratumoral ratio, cDC1 numbers (correlated with CD8+ T cell percentages) and cDC2 numbers (correlated with CD4+ T cell percentages) together correlated with response. Krummel closed with a description of T cell/myeloid cell “archetypes”, which could be used to predict responsiveness to checkpoint therapies.
In trying to identify the differences between T cell-inflamed and non-inflamed melanomas, Stefani Spranger discovered that the WNT/β-catenin pathway was upregulated in the latter. Digging further, she found that β-catenin-expressing tumors lacked tumor-resident, Batf3-driven, CD103+ cross-presenting DCs, which were the predominant source of CXCR3 ligands (CXCL9 and CXCL10) and were required for the recruitment and priming of effector T cells. To further clarify the role of tumor-resident DCs, Spranger analyzed models of regressing and progressing tumors. She found that the number of cross-presenting DCs increased over time after tumor implantation in regressing, but not in progressing tumors, and this was accompanied by an increase in CD8+ T cells. However, in regressing tumors, deletion of Batf3 or Clec9A showed that the T cell response was independent of conventional cross-presenting cDC1 DCs, begging the question of what type of DCs were involved in the antitumor response. Single-cell RNAseq analysis revealed a new subset of DC-like cells that were only present in regressing, but not in progressing tumors. These cells were able to cross-present and prime CD8+ T cells, but with different kinetics than cDC1s. Based on these results, Spranger generated a working hypothesis that multiple subsets of tumor-resident DCs (including non-canonical DCs) with cross-presenting capabilities are necessary for an effective immune response and speculated, based on the Clec9A-/- results, that the antigen source for such DCs was not apoptotic cells.
Noor Momin explored the hypothesis that anchoring cytokines to the abundant and long-lived collagen in the tumor would optimize local efficacy while reducing systemic toxicity. The anchoring domain used was Lumican, a natural leucine-rich repeat mouse (and human) protein with high affinity for Type I and IV collagen. When IL-2 was fused to the C-terminus of Lumican and injected intratumorally (i.t.) in a B16F10 mouse tumor model together with systemic delivery of TA99, an antibody targeting TYRP-1 on the surface of B16F10 cells, nearly 100% of the mice were cured – significantly higher than the cure rate with intratumor non-anchored IL-2. The treated mice also developed vitiligo, a marker of an anti-melanocyte response. Extending this concept to other cytokines, a fusion of a single-chain IL-12 to Lumican (separated by a mouse serum albumin [MSA] linker) dramatically improved the efficacy of CAR T cells in the B16F10 model. To examine systemic toxicity, i.t. injection of Lumican-MSA-IL-12 was compared to i.t. or intraperitoneal (i.p.) injection of MSA-IL-12. I.t. or i.p. injection of MSA-IL-12 both resulted in significant and comparable weight loss, while i.t. injection of Lumican-MSA-IL-12 did not. In the metastatic 4T1-LUC tumor model, Lumican-MSA-IL-12 together with anti-PD-1 controlled distal tumors. Finally, the Lumican-MSA-IL-12 fusion was also active in EMT6, MC38, and tamoxifen-induced Braf/Pten genetically-engineered mouse models, demonstrating wide applicability.
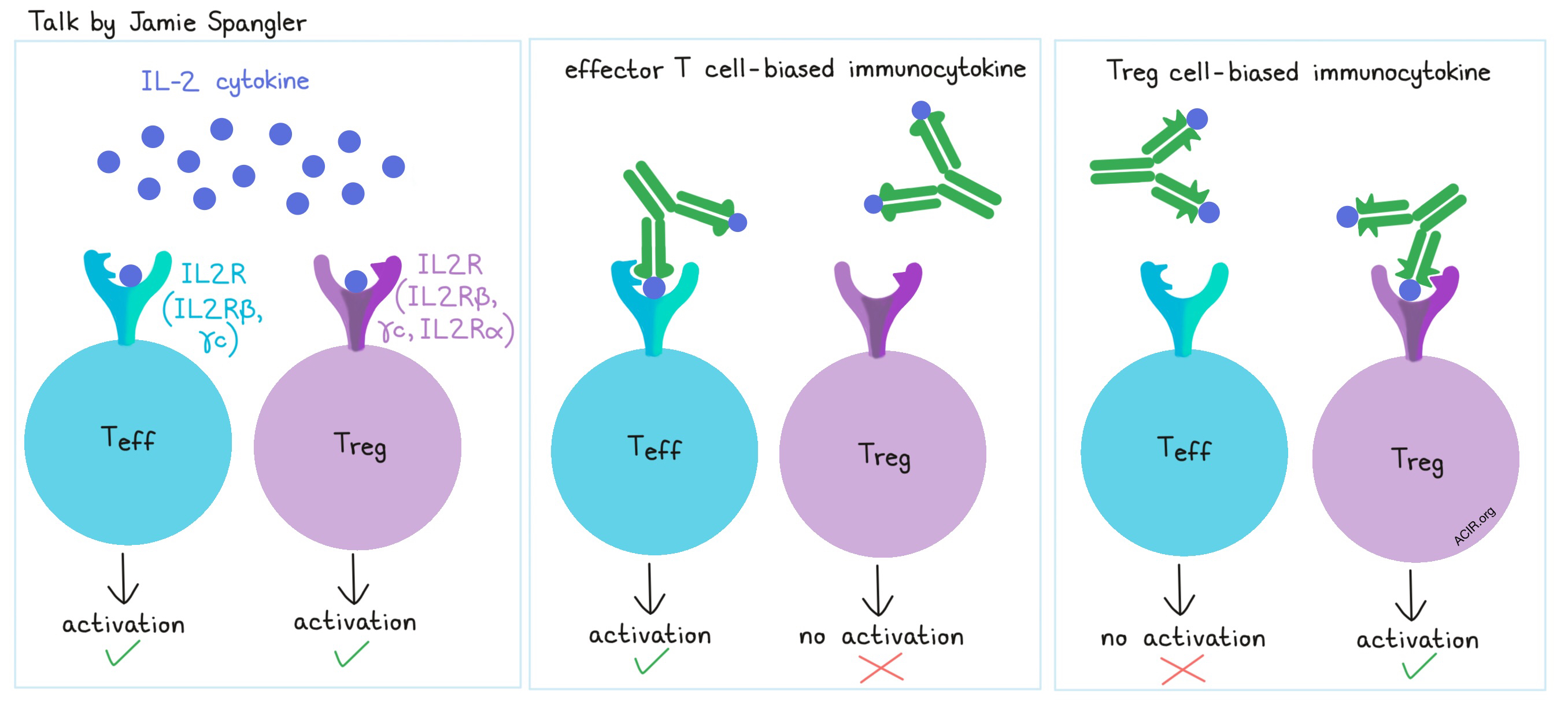
Discussing strategies to improve the use of cytokines in the clinic, Jamie Spangler described her team’s efforts to use molecular engineering to improve IL-2 cytokine therapy. IL-2 is used to stimulate the IL-2 receptor (composed of IL2Rβ and γc) on effector T cells, however, it also targets the IL-2 receptor (composed of IL2Rβ, IL2Rα, and γc) on regulatory T cells, which ultimately hinders its therapeutic efficacy. Spangler’s group took aim at limiting this pleiotropic effect by identifying antibodies that obstruct IL-2 from interacting with certain portions of the receptors, thereby directing IL-2 towards either effector or regulatory T cells. Based on their structural understanding of the interactions, the researchers engineered an effector cell-biased immunocytokine, which preferentially targeted effector cells over Tregs, and overall enhanced the antitumor effect of IL-2. In separate work, Spangler and colleagues also generated a computationally designed, hyper-stable, effector cell-biased IL-2, which induced superior antitumor immunity in multiple tumor models compared to the natural IL-2 cytokine. In addition to having an extended half-life, the hyper-stable IL-2 reduced toxicity and showed no evidence of immunogenicity. A similar approach led to the design of a hyper-stable IL-4, which enhanced macrophage M2 polarization and CD4+ T cell Th2 polarization.
Yvonne Chen began by discussing the use of dual-targeted CD19/CD20 “OR-Gate” CAR T cells (that express two individual chimeric receptors each targeting CD19 or CD20) to reduce the likelihood of tumor immune escape due to antigen loss. In mouse models with wild-type Raji tumors (CD19+/CD20+), CD19-targeted CAR T cells or CD19/CD20 “OR-Gate” CAR T cells both initially cleared tumors, but the OR-Gate CAR T cells were more effective in preventing late relapse. In mice with mixed Raji tumors (75% CD19+/CD20+, 25% CD19-/CD20+), only the OR-Gate CAR T cells effectively cleared tumors, with no observed late relapses. Additional experiments demonstrated that compared to dual CARs, bispecific CARs (two targeting domains linked together on the same receptor) were more efficiently produced and expressed on the cell surface (due to the smaller genetic footprint and increased transduction efficiency) and showed more efficient antigen-stimulated proliferation. Chen’s lab also generated bispecific BCMA/CS1-targeting CAR T cells for multiple myeloma, which outperformed single-input CAR T cells in a mouse model with variable antigen presentation. In the second half of the talk, Chen discussed the possibility of rewiring the T cell response to resist endogenous TGFβ signaling in the TME and simultaneously use TGFβ as an activating signal instead. To do this, the researchers constructed a CAR with a TGFβ-specific single-chain antibody fragment. The researchers also took advantage of the natural dimeric structure of TGFβ, which allowed the CAR construct to signal through receptor dimerization following binding to TGFβ, causing the engineered cells to respond to TGFβ as if it were a growth-promoting cytokine.
Michael Birnbaum discussed ways of better understanding T cell recognition of target antigens. Currently, the database of all MHC-interacting peptides is variable between alleles and limited for many, so in an effort to generate large amounts of unbiased data, Birnbaum and his colleagues created a cleavable pMHC yeast display system, consisting of a genetically encoded library of highly random potential peptide binding sequences linked to an MHC II molecule and separated by a protease cleavage site. Expression of the construct in yeast leads to a collection of single yeast cells, each with a cell surface MHC II containing an encoded peptide. Following protease cleavage to free the peptide from MHC II, and the addition of the chaperone non-canonical MHC II molecule HLA-DM to accelerate the loss of low-affinity peptides, yeast-containing stably-bound peptides can be isolated. After multiple rounds of enrichment, amino acid preferences and structural relationships important to the peptide:MHC II interaction can be determined. Using this system, the researchers identified massive amounts of peptide hits – data on which they could potentially train a peptide prediction algorithm. Applying this approach could identify motifs across the broad repertoire of HLA polymorphisms. Birnbaum’s current strategies are limited to prediction for MHC II, but his team is interested in doing similar analysis for MHC I. He also showed it was possible to use his system to potentially exploit HLA-E, which has a more diverse peptide repertoire than previously thought and can be recognized by the adaptive immune system. Finally, Birnbaum discussed use of the “MAD-HYPE” multi-cell per well TCR-sequencing and de-convolution approach to extract T cell receptor information and identify both abundant and rare T cell clones.
Glenn Dranoff described the approach at Novartis to conduct small, data-rich exploratory clinical trials, particularly for potentially complementing therapies. He reviewed some of their early clinical trial results with an emphasis on correlative analysis. Trials of CD19-targeting CAR T cells to target B cell malignancies show that many patients achieve complete – though not always durable – responses, and about half of patients derive long-term benefits. Adverse events and tumor immune escape due to antigen loss remain hurdles to overcome. Dranoff next discussed preliminary efficacy data for a non-FcγR binding CD123 x CD3 bispecific antibody (allowing T cell-mediated killing of CD123-expressing cells regardless of T cell specificity) which was designed to be dosed intermittently. In a trial of 18 AML patients, the antibody reduced bone marrow blasts in 56% of patients, with 28% of patients achieving an objective (CR or CRi) response. Trials of PD-1 blockade in patients with anaplastic thyroid carcinoma and PD-L1 blockade in patients with chordoma and alveolar soft parts sarcoma showed increases in CD8+ T cell infiltration and inflammatory cytokines in the TME; patients with otherwise refractory disease achieved objective responses. The combined blockade of LAG3 and PD-1 showed preliminary activity against triple-negative breast cancer with appropriate changes in T cell signatures. Dranoff also turned attention to the potential modulation of myeloid cells by targeting TIM3, which is expressed on myeloid cells and may serve as a regulator of dendritic cell function. Finally, Dranoff discussed how NIR178, an A2AR inhibitor that targets the immunosuppressive adenosine pathway, showed significant single-agent activity in a multi-tumor type trial and could be combined with anti-PD-1.
by Lauren Hitchings, Anna Scherer, and Ed Fritsch



