Cancers are the escape artists of the biological world, each with a unique act by which it eludes or overpowers the immune system of an affected organism. Some human cancers have been found to constitutively express indoleamine 2,3-dioxygenase 1 (IDO1), which rapidly catabolizes tryptophan to kynurenine, creating an immunosuppressive microenvironment that prevents T cell infiltration and proliferation – the classic “cold” tumor. In this study, Hennequart et al. sought to elucidate the mechanism behind constitutive IDO1 expression as an immune escape method of human cancer.
Hennequart et al. performed a series of studies using a BRAF-mutated human melanoma cell line that constitutively expresses IDO1 to study the various pathways involved in this unique model of immune escape. They first confirmed that constitutive IDO1 expression was not caused by the IFNγ signaling pathway that can induce IDO1 expression in almost any tissue, then moved on to instead explore the contributions of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2), the latter of which is known to play a role in normal IDO1 induction in dendritic cells.
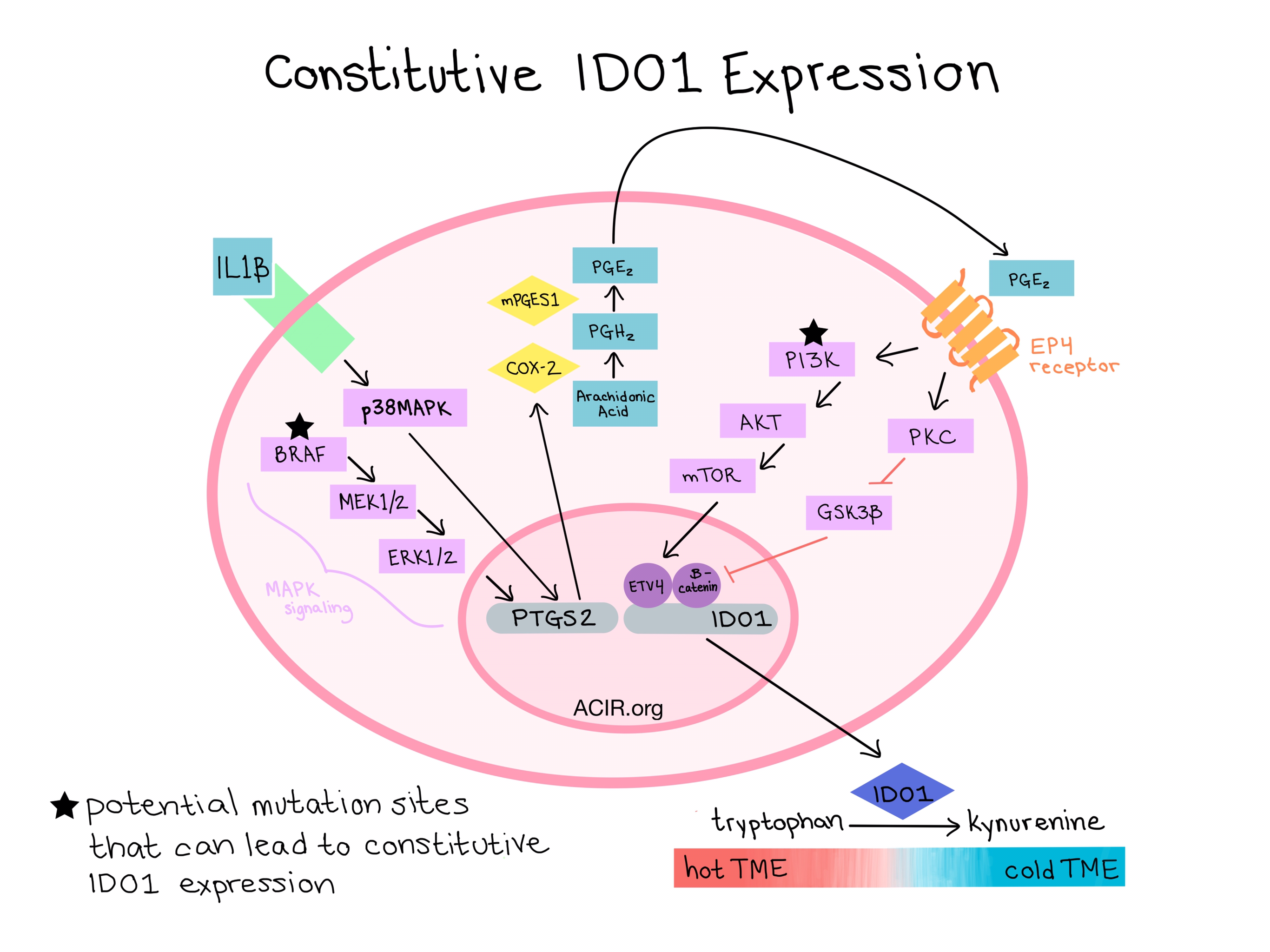
The proposed mechanism underlying constitutive IDO1 expression is as follows:
- MAPK signaling (BRAF → MEK1/2 → ERK1/2) and/or an autocrine loop of IL1β signaling through the MAPK pathway help to support constitutive expression of COX-2 and mPGES1.
- COX-2 expression leads to sustained PGE2 production.
- PGE2 is recognized by EP4 receptors, triggering two separate signaling pathways that induce IDO1 transcription.
- In the PI3K signaling pathway, PI3K activates AKT, which activates mTOR. mTOR activation likely depends on the transcription factor ETV4, which functions downstream and binds to the IDO1 promoter.
- In the PKC signaling pathway, PKC deactivates GSK3β. Active GSK3β typically phosphorylates β-catenin, which leads to its degradation. Thus, deactivation of GSK3β leads to an excess of activated β-catenin, which binds to and activates the IDO1 promoter.
- IDO1 expression leads to degradation of tryptophan into kynurenine.
- The low tryptophan:kynurenine ratio creates an immunosuppressive microenvironment in which T cells cannot survive or proliferate.
The researchers confirmed that the COX-2/PGE2 pathway is responsible for constitutive IDO1 expression in a wide range of human cancer types including cholangiocarcinoma, head and neck carcinoma, sarcoma, ovarian carcinoma, non-small cell lung carcinoma, and another melanoma. Most of these IDO1-expressing tumor cell lines had mutations in genes from the PI3K or MAPK pathways, or in the receptors that lead to the activation of these pathways. Despite previous reports that STAT3 affects IDO1 production, Hennequart et al. found no direct role of STAT3.
To determine whether they could induce an anti-tumor immune response by targeting the COX-2 pathway to knock down constitutive IDO1 expression, Hennequart et al. used a human ovarian cancer line (SCOV3) that is sensitive to the COX-2 inhibitor, celecoxib. Human tumor xenografts were controlled in mice that received both celecoxib and allogeneic lymphocyte therapies, but not either alone. The same tumors showed reduced IDO1 which likely allowed for improved lymphocyte infiltration and proliferation in tumors that were otherwise “cold” in control mice and mice treated with celecoxib alone. Overall, results from the combination immunotherapy experiments further confirmed the mechanism by which celecoxib induced tumor rejection through repression of IDO1.
These mechanistic studies reveal additional approaches (COX-2 inhibition with the approved drug celecoxib or inhibitors of the PIK3K pathway) to overcome the “non-inflamed” tumor microenvironment characteristic of “cold” tumors caused by constitutive IDO1 expression. Such approaches may be additive or synergistic with the inhibitors of IDO1, such as epacadostat, currently being examined in the the clinic.
by Lauren Hitchings



