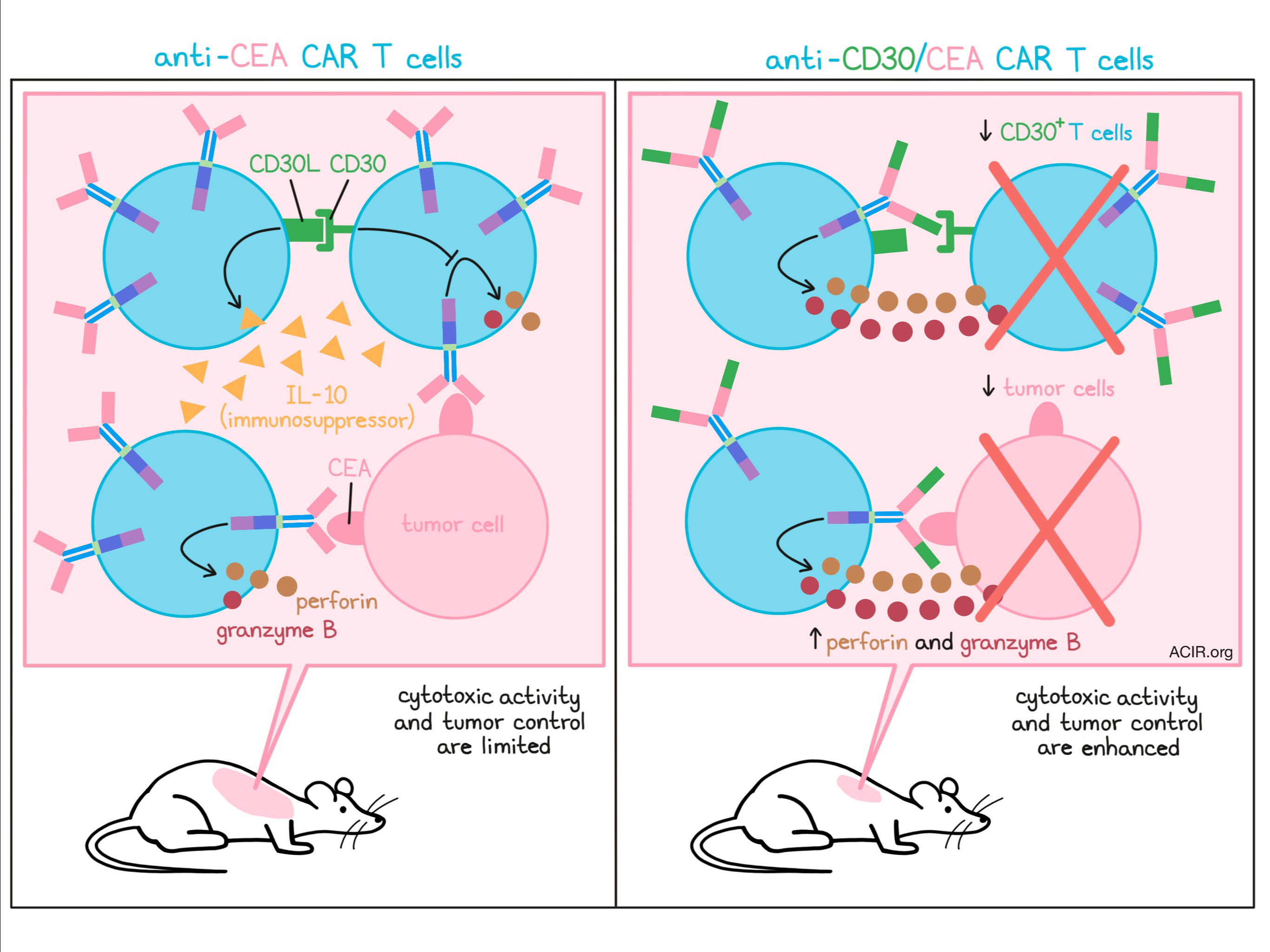
In the hunt to expand the arsenal of checkpoint regulators, Hombach et al. investigated the possibility of targeting the CD30-CD30L axis. CD30 and/or CD30L can be expressed on a number of immune cells (T cells, B cells, and some APCs), and prior evidence has suggested that it may play a role in limiting T cell activation. While CD30 has previously been targeted as a tumor antigen on hematological malignancies, its transient expression on healthy T cells upon activation may also serve as a useful immunomodulatory target. In a study recently published in Molecular Therapy, Hombach et al. explored the role of CD30 in T cell activation and designed a dual-specificity CAR T cell that targets a specific antigen on tumors, while targeting CD30 within the T cell product.
To begin, Hombach et al. investigated whether CD30 acts as a checkpoint upon primary T cell activation. Using an anti-CD30 immunotoxin or anti-CD30 CAR T cells, the researchers effectively eliminated a sub-population of CD30+ cells from carcinoembryonic antigen (CEA)-targeted CAR T cells. Compared to control anti-CEA CAR T cells, anti-CEA CAR T cells depleted of the CD30+ population showed enhanced antitumor activity against CEA+CD30- cancer cells in vitro, suggesting that CD30+ cells limit the efficacy of the final CAR T cell product. Anti-CEA CAR T cells depleted of CD30+ cells also produced less of the immunosuppressor IL-10. Since IL-10 was primarily secreted by CD30L+ T cells, reduced IL-10 upon CD30 blockade indicates that CD30 likely regulates IL-10 production through CD30L signaling. Production of IL-2 and IFNγ were not affected.
Upon primary activation, CD30L and CD30 are known to be rapidly and transiently upregulated, and in this study, researchers observed similar expression of CD30 upon secondary activation of previously activated T cells. They also found that in cocultures of peripheral blood lymphocytes (PBLs) that had not been previously activated and PBLs that had been previously activated, simultaneous primary and secondary T cell activation resulted in the highest increase in CD30+ T cell populations. To determine the role of CD30 in activation, the researchers showed that high CD30 expression was associated with reduced cytotoxicity in anti-CEA CAR T cells. Treatment with HRS4, a CD30-blocking mAb, improved the cytotoxic activity of anti-CEA CAR T cells and reduced their production of IL-10.
In an effort to utilize CD30 blockade to enhance CAR T cell activation and improve effector functions, Hombach et al. designed a novel CAR containing both a CEA-targeting domain and a CD30-targeting domain (scFv derived from the CD30-blocking HRS3 mAb). This dual-specificity CAR was expressed at slightly lower levels on the T cell surface compared to anti-CEA CARs, however, it still effectively reduced the portion of CD30+ T cells within the population. Anti-CD30/CEA CAR T cells effectively eliminated both CD30+CEA- and CD30-CEA+ tumor cells in vitro. Further, the dual-specificity CAR outperformed anti-CEA CAR T cells against CEA+ tumor cells, while the anti-CD30 CAR T cells and anti-CD30/CEA CAR T cells demonstrated comparable killing of CEA-CD30+ tumor cells. In vivo, anti-CD30/CEA CAR T cells were more effective than anti-CEA CAR T cells in preventing the establishment of aggressive CEA+ tumors.
Investigating the mode of action by which the anti-CD30/CEA CAR enhances T cell efficacy over anti-CEA CAR T cells, the researchers first determined that the enhanced cytotoxicity conferred by the dual-specificity CAR was not due to differential surface expression by either CD4+ or CD8+ T cells, nor due to differential expansion of either subset. Both CD4+ and CD8+ T cells exhibited improved cytotoxic activity against CEA+ tumor cells when transduced with the dual-specificity anti-CD30/CEA CAR compared to the single-specificity anti-CEA CAR. Dual-specificity CAR T cells also produced higher levels of perforin and much higher levels of granzyme B than anti-CEA CAR T cells.
To better understand how the anti-CD30 domain behaves within the dual-specificity CAR, the researchers replaced the HRS3-derived anti-CD30 scFv with an anti-CD30 scFv derived from the agonistic SH313B5 antibody. Expression of the anti-CD30(SH313B5)/CEA CAR was comparable to the anti-CD30(HRS3)/CEA CAR, however, it did not reduce the CD30+ T cell population. Furthermore, anti-CD30(SH313B5)/CEA CAR T cells were less cytotoxic than anti-CEA CAR T cells, suggesting that when integrated into a CAR design, the HRS3-derived anti-CD30 scFv blocked CD30, while the SH313B5-derived scFv agonized CD30, potentiating its negative signaling.
Next, the researchers explored whether targeting CD30 was comparable to targeting other activation-associated makers by replacing the CD30-targeting domain of the dual-specificity CAR with a domain targeting CD25 (a component of the IL-2 receptor that is upregulated in T cells upon activation). Compared to T cells expressing the anti-CD30/CEA CAR, T cells expressing the anti-CD25/CEA CAR showed more rapid expansion and similar persistence, but did not exhibit enhanced CEA-directed cytotoxic activity nor increased production of perforin or granzyme B.
Finally, to test whether the CD30 blocking domain could be integrated into CAR T cells with other antigen targets, the researchers developed an anti-CD30/TAG72 CAR. Similar to the anti-CD30/CEA CAR, surface expression following transduction was lower than anti-TAG72 CAR expression, but the dual-specificity CAR reduced CD30 levels and more efficiently targeted TAG72+ tumors compared to single-specificity TAG72 CAR T cells.
Overall the researchers showed that T cell expression of CD30 upon activation limits effector functions while enhancing production of the suppressive cytokine IL-10. They found that blocking CD30 may be an effective way to reverse such effects, and that this could be achieved in CAR T cells by integrating a CD30-blocking domain into a tumor-antigen targeting CAR. This novel CAR design could be used to target a broad range of tumor antigens while simultaneously neutralizing the activation-limiting effects of CD30 on T cells and enhancing antitumor activity.
by Lauren Hitchings
MEET THE RESEARCHER
This week, lead author Hinrich Abken answered our questions.

What prompted you to tackle this research question?
It is well established that specifically redirected T cells can control cancer in the long-term. Spectacular examples are chimeric antigen receptor (CAR) engineered T cells that recognize and destroy cancer cells upon binding to a cognate antigen on the surface of cancer cells. While the strategy is very efficacious in the treatment of leukemia and lymphoma, CAR T cells mostly fail in the treatment of solid cancer, raising the need for sustaining T cell activation. We therefore looked for key regulators of T cell activation beyond classical checkpoints and asked how to combine the “modulation of the regulator” with a CAR-initiated T cell response.
Physiologically, CD30 is involved in the initiation of a productive response of mature T cells, however, it is tightly regulated by reciprocal signaling through CD30L during the entire process. Transiently expressed by cells of the innate and adaptive immune system, CD30L interacts with freshly activated CD30+ T cells to reduce the level of activation. In order to prolong T cell activation early after initiation we asked whether CD30 targeting positively affects CAR T cell immunity, in particular during a CAR-redirected attack against cancer cells that lack CD30.
What was the most surprising finding of this study for you?
While targeting CEA+CD30- cancer cells of a gastrointestinal carcinoma by anti-CEA CAR T cells we noticed that eliminating CD30+ cells from the T cell pool substantially increased the anti-cancer activity of CAR T cells. This could be done by adding a CD30-redirected toxin or, notably, by adding T cells redirected by a CAR against CD30+ T cells. The anti-CEA CAR T cell population “cleaned” of CD30+ T cells was more efficacious than the population containing CD30+ T cells. This opened the doors to combine anti-CD30 and anti-cancer activity into one CAR to make more powerful T cells.
What was the coolest thing you’ve learned (about) recently outside of work?
That science and arts are connected: good science is an art and good arts are a science.



