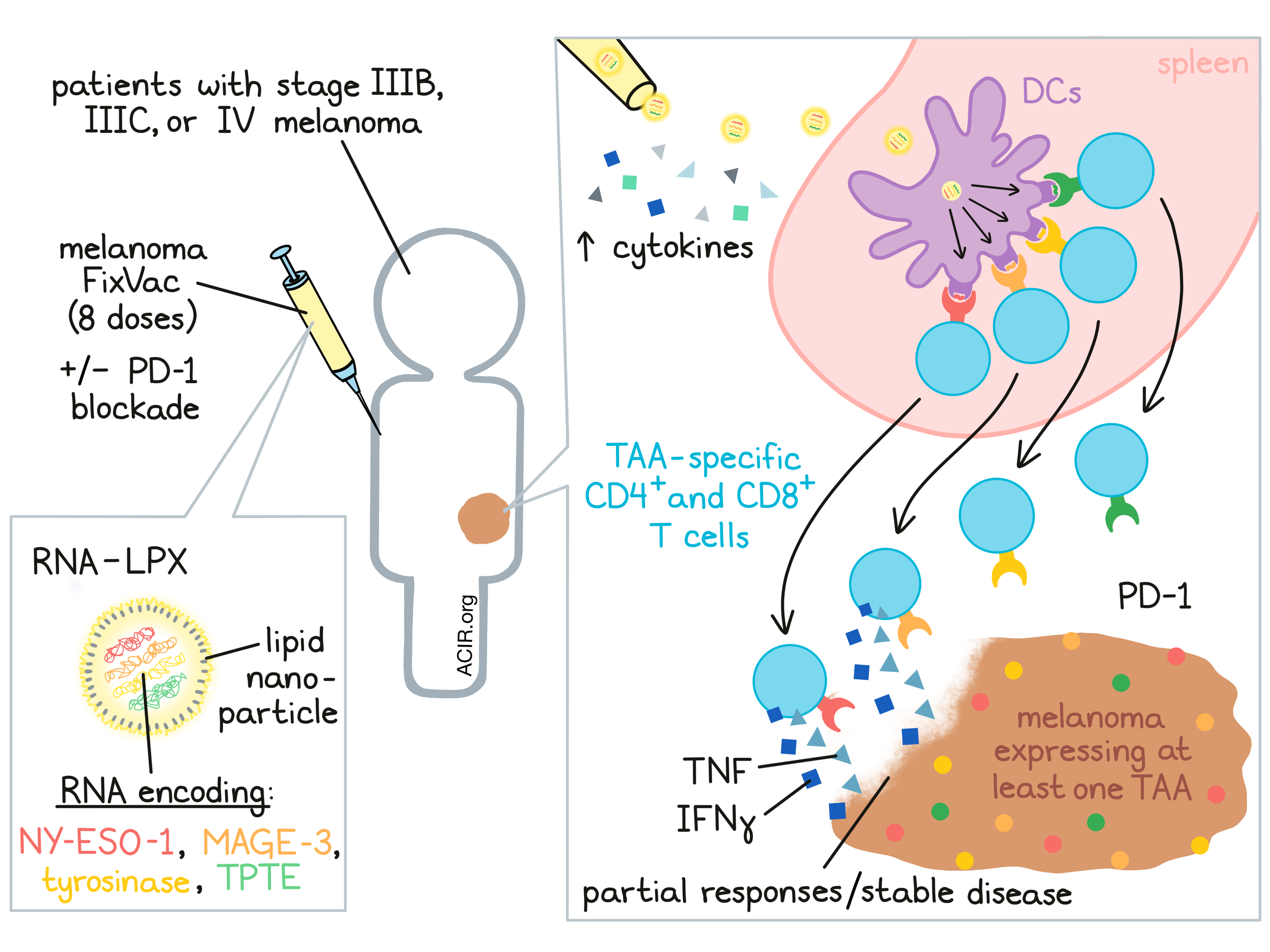
In the world of cancer vaccines, targeting tumor-associated antigens (TAAs) has historically yielded only weak immune responses. In an effort to improve vaccine targeting of TAAs, researchers developed a nanoparticulate liposomal RNA (RNA-LPX) that targeted dendritic cells in vivo and induced antigen presentation on HLA-I and HLA-II molecules in lymphoid compartments in mice. Along with costimulation through TLR-mediated type 1 IFN-driven inflammatory mechanisms, this vaccine induced expansion of antigen-specific T cells. Following studies showing that this strategy could effectively induce strong, durable antitumor responses in mice, Sahin et al. initiated a phase I clinical trial, the results of which were recently published in Nature.
The phase I clinical trial was initiated as a multicenter, open-label, dose-escalation trial to evaluate the safety and tolerability of the RNA-LPX vaccine FixVac. Melanoma FixVac encoded four TAAs – NY-ESO-1, MAGE-A3, tyrosinase, and TPTE – which are commonly expressed in melanomas, are immunogenic, and show restricted expression in normal tissues. FixVac was delivered intravenously as an RNA-LPX. Patients with stage III B or C, or stage IV melanoma were treated with FixVac or with a combination of FixVac and a clinically approved PD-1 checkpoint blockade. The 89 patients enrolled in the trial had tumors that expressed at least one of the TAAs encoded in the vaccine, but were not HLA-selected. Patients underwent eight vaccinations in a prime/boost protocol, followed by optional continued monthly treatment, with some patients receiving dose escalation up to their target dose.
Interrogating the efficacy of FixVac targeting to lymphatic tissue, the researchers identified increased metabolic activity, specifically in spleen, shortly after vaccine administration. This served as a marker of TLR ligand stimulation, indicating fast targeting and activation of immune cells in lymphoid organs. Assessing adjuvanticity, the researchers measured levels of cytokines in plasma and found that IFNα, IFNγ, IL-6, IP-10 and the IL-12 subunit p70 all transiently increased with each FixVac dose. Interestingly, RNA-LPX triggered cytokine release at much lower concentrations in humans than in mice. Along with cytokine release, vaccine doses induced transient increases in body temperature. Adverse events consisted mainly of mild to moderate flu-like symptoms, which generally occurred early on, could be managed with antipyretics, and resolved within 24 hours.
Next, the researchers investigated the immunogenicity of melanoma FixVac by performing an ex vivo IFN-γ ELISpot on samples of bulk PBMCs, CD4+ T cell-depleted PBMCs, and CD8+ T cell-depleted PBMCs taken from 50 patients before and after 8 injections of the vaccine. The samples were incubated with overlapping peptides representing full-length sequences of TAAs. Based on this analysis, more than 75% of patients showed ex vivo responses to at least one TAA, and the highest magnitude responses were by CD8+ T cells. Further, samples from 20 patients were analyzed using post-IVS IFN-γ ELISpot, in which autologous dendritic cells were loaded with TAAs to serve as targets. In this analysis, samples from all 20 patients showed a T cell response against at least one TAA. Most responses involved CD4+ T cells or both CD4+ and CD8+ T cells. Most of the responses that occurred were de novo responses.
To further evaluate de novo CD8+ T cell responses ex vivo, Sahin et al. used HLA multimer analysis and intracellular cytokine staining and found that antigen-specific T cells ramped up within 4-8 weeks and were of an effector memory phenotype. These cells expressed PD-1 and secreted IFN-γ and TNF upon antigen-specific restimulation. Most patients had CD8+ T cell responses to multiple epitopes. In patients who underwent monthly maintenance vaccinations, TAA-specific T cell populations continued to expand or remained stable long term, and memory phenotypes were present. Further, when TAA-specific TCRs from vaccine-expanded T cells were transfected into healthy donor T cells, they effectively killed TAA-expressing melanoma cells.
While clinical activity was not a primary endpoint of this trial, Sahin et al. did perform a preliminary analysis of the clinical activity of FixVac by assessing the best objective response in 42 patients. Almost all of these of the patients had stage IV disease and had already been exposed to one or more checkpoint blockade therapies. In 25 patients receiving FixVac monotherapy, 3 experienced a partial response and 7 achieved stable disease. Another showed a complete metabolic remission of their metastatic lesions. In 17 patients treated with FixVac and anti-PD-1, 6 developed a partial response. Regression of target lesions occurred across all doses, and objective response correlated with baseline tumor burdens. Most patients who experienced a partial response or stable disease showed durable disease control for up to two years of observation.
For five patients who had partial responses, the researchers had sufficient blood samples for detailed characterization of immune responses. Analysis of these patients revealed strong responses, mostly directed to NY-ESO-1 and MAGE-A3. It also showed that some patients treated with FixVac became re-sensitized to checkpoint blockades, likely due to the de novo expansion of TAA-specific T cells expressing PD-1.
Based on the results of this clinical trial, Sahin et al. determined that FixVac induced a transient cytokine response, antigen presentation by DCs, and strong CD8+ and CD4+ T cell responses to the encoded TAAs. The prime/boost protocol expanded the pool of circulating antigen-specific T cells, particularly those targeting NY-ESO-1 and MAGE-A3, that were functional, recognized their target epitopes on melanoma, and exhibited strong cytolytic activity. Continued vaccination appeared to maintain these cell populations, and patients with strong, diversified T cell responses seemed to have better objective responses. FixVac also seemed to synergize with PD-1 checkpoint blockade, and could re-sensitize patients to checkpoint blockade, even when they had previously been resistant, aligning with the observation that T cells induced by melanoma FixVac expressed PD-1. The results of this study validate the possibility of inducing potent immune responses to non-mutated, shared TAAs using cancer vaccines, which may be particularly useful against cancers without high tumor mutation burdens or strong neoantigens to target.
by Lauren Hitchings



