Even though MAPK inhibitors (MAPKi, such as BRAFi or MEKi) can be an effective treatment strategy, therapy resistance limits their benefits. However, in clinical studies, patients with BRAFV600E/K metastatic melanoma, who had received prior treatment with checkpoint inhibitors, had increased efficacy of MAPKi with longer progression-free survival. Therefore, Wang, Liu, Yang, and Algazi, et al. investigated in various mouse models, including melanoma and melanoma with brain metastasis, how pre-treatment with anti-PD-L1 affects MEKi therapy efficacy. Their results were recently published in Cancer Cell.
The researchers used syngeneic subcutaneous tumor models to assess whether a brief pretreatment with anti-PD-L1 or anti-PD-1 (lead-in) could improve responses to subsequent MEKi therapy, with or without continuous PD-L1 blockade. Dosing started after tumors were established (d0), and PD-L1 inhibition was initiated at d0 or d7 and given continuously thereafter, or was given only between d0 and d7, while the MEKi Trametinib was given continuously starting at d0 or d7. The tests were performed in six different models: BrafV600E melanoma with a high mutational burden (YUMM1.7ER), NrasQ61R melanoma (NIL), NrasQ61R melanoma with a high mutational load (NILER1-4), Nf1-/- melanoma (mSK-Mel254), KrasG12C colorectal carcinoma (CT26), and KrasG12C pancreatic adenocarcinoma (KPC). In these models, treatment with only anti-PD-L1 had limited to no effects. MEKi alone or followed by PD-L1 also had limited effects, as did combination treatment initiated simultaneously. However, most tumor regression was seen in mice treated with anti-PD-L1 lead-in followed by MEKi. Using one cell line (mSK-Mel254), anti-PD-1 was assessed similarly, and also for that checkpoint inhibition strategy, the lead-in followed by MEKi was most effective. Combining BRAFi and MEKi with anti-PD-L1 also resulted in the best response when anti-PD-L1 was given as a lead-in.
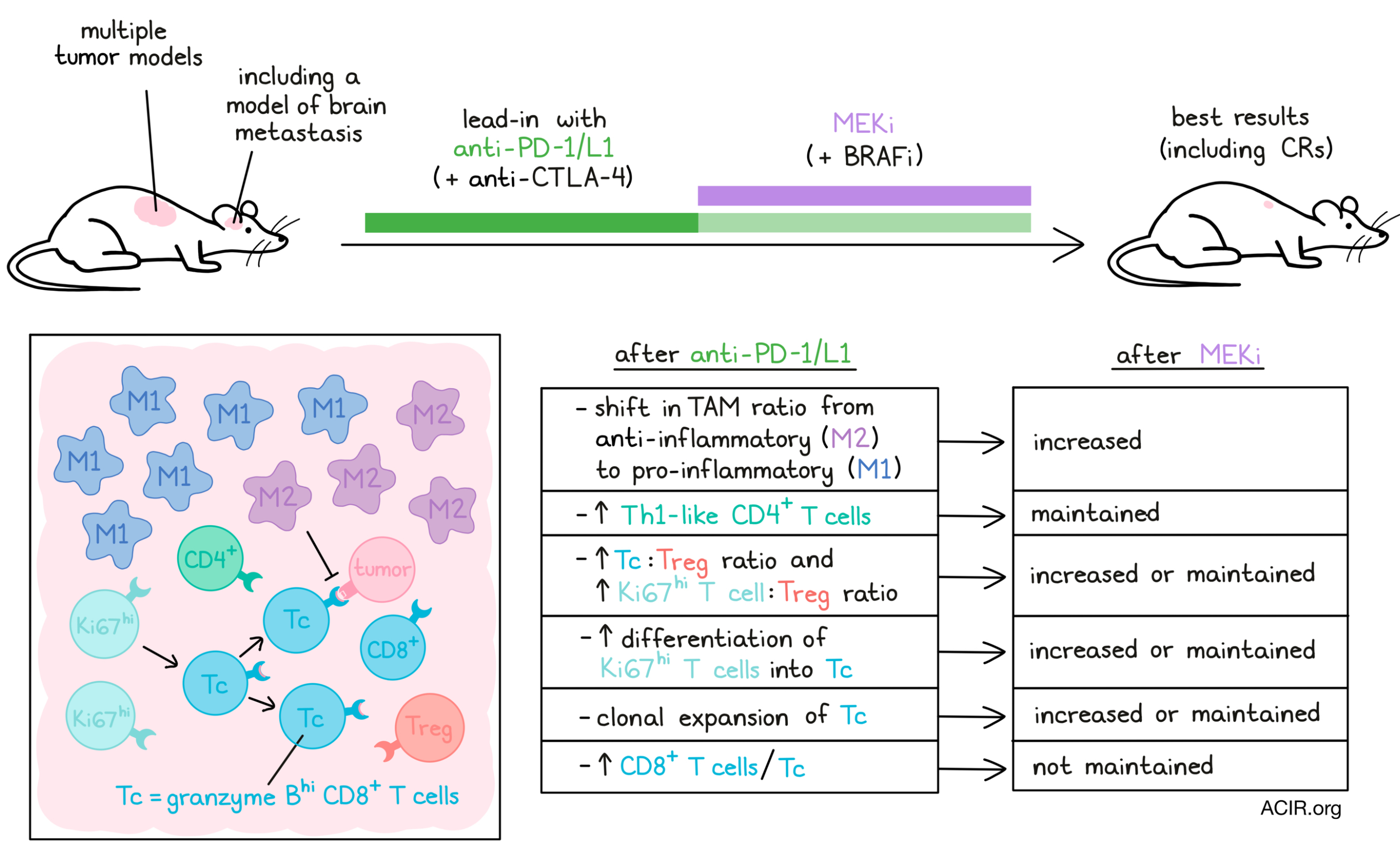
To explore mechanisms, the effects on immune populations in the tumor microenvironment were assessed by CyTOF analyses on d7 (after lead-in) and d10 and d14 (both after MEKi). The CD45+ population varied widely by model and over time after the different treatments. In all mice, the tumor-associated macrophages (TAMs) were the most abundant immune subsets, but there was an increase in iNOS+ M1-like TAMs only in the anti-PD-L1 lead-in therapy regimens. Further evaluating TAMs at the transcriptomic level using single-cell RNAseq revealed that the pro-inflammatory macrophages became more abundant than the anti-inflammatory TAMs in mice treated with the lead-in anti-PD-L1 treatment, which was further enhanced by MEKi on day 7, but not the other MEKi treatment regimens.
CD8+ T cells increased after anti-PD-L1 only in the YUMM1.7ER model, and increases in this population after MEKi treatments were higher in the YUMM1.7ER and CT26 models; however, these increases did not correlate with therapeutic efficacy. After lead-in therapy, Th1-Like CD4+ T cells increased, and anti-PD-L1 treatment also increased Granzyme BhiCD8+ T cells (Tc), but this CD8+ T cell increase was not maintained after MEKi.
The T cell populations were further analyzed by coupled scRNAseq and scTCRseq. A total of 1,384 clonal T cells were found, and the CD8+ Tc population had the highest degree of clonal expansion and concentration of clonal cells. Transition index analyses suggested a differentiation trajectory from proliferative Ki-67hiCD8+ T cells to the clonally expanded Tc population. Ratios of these populations versus Tregs were increased after lead-in of anti-PD-L1 and were maintained or further increased after the MEKi treatment. In contrast, low ratios were detected after the other treatment regimens. The lead-in treatment followed by MEKi on day 7 also increased transition from Ki-67hiCD8+ to Tc subsets, which was not seen in other treatment groups. Both T cell subsets expressed Ifnγ, and the Tc subsets upregulated activation, inhibitory, and effector genes, supporting their role in antitumor responses.
The researchers next assessed the critical roles of various populations in the antitumor effects of the lead-in strategy followed by MEKi. Targeting the M2-like TAMs with a CD206 agonist had little impact on its own, but in the model treatment with lead-in PD-L1 followed by MEKi, on d7 an increased and more durable tumor regression was achieved, suggestive of improved priming. Neutralization of CD8+ T cells eliminated the beneficial effects of this most efficacious treatment schedule, revealing CD8+ T cells as critical for the antitumor effects.
Therapy resistance to MAPKi is a particular issue in melanoma brain metastasis. Therefore, the researchers assessed whether the lead-in strategy would also increase efficacy in this disease setting. Using BrafV600E melanoma cells, tumors in the brain and other organs were initiated by intracardial injection. These mice were subjected to the various treatment regimens, defining d0 as the day of initiation of MEKi therapy, when intracranial tumors were established. Treatment with MEKi alone reduced tumor burden, but with limited survival benefit. Treatment of anti-PD-L1 at day 0, when MEKi treatment initiated, had no effects, but treatment with anti-PD-L1 four days earlier (d-4) followed by MEKi on day 0 suppressed tumor growth at both intracranial and extracranial sites and extended survival. Therefore, in this model, two doses of anti-PD-L1 before combining it with MEKi resulted in higher efficacy and delayed MEKi resistance. The two best responding regimens (anti-PD-L1 on d-4 and d0 of MEKi) were further analyzed by replacing MEKi with BRAFi and MEKi. The mice treated with the lead-in PD-L1 survived longer than those that received PD-L1 blockade simultaneously with the MEKi and BRAFi. When anti-PD-1 was substituted for anti-PD-L1 in the most efficacious treatment regimen (anti-PD-L1 on d-4 and BRAFi and MEKi on d0), the efficacy was higher than anti-PD-1 alone. Combining anti-PD-L1 with anti-CTLA-4 further improved the efficacy. In ovarian and brain tumors the T cell clonotypes were assessed at d0 and d3 of the various treatment strategies. In the tumors treated with anti-PD-L1 four days before MEKi, T cell clonality was higher, and TCR diversity was lower on day 3 after MEKi initiation, with the highest concentration of large TCR clones.
These data suggest that pretreatment with anti-PD-1 axis therapy before initiation of MEKi in multiple primary and metastatic tumor types, including melanoma brain metastases, may synergistically increase anti-PD-1 axis efficacy and delay MEKi resistance, with a potentially increased benefit when checkpoint blockade targeting the PD-1/PD-L1 axis is combined with blockade of CTLA-4.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, senior authors Roger Lo and Gatien Moriceau answered our questions.

What prompted you to tackle this research question?
RL: Therapies targeting the MAPK and PD-1/L1 cancer pathways have revolutionized oncology. Despite having two revolutionary ways to treat cancer developed during the last decade (first in melanoma), many patients still go from one form of treatment to another and ultimately succumb to their disease. We had clues from prior studies that these two forms of therapy, when applied individually until they fail the patients, may create cross-tolerance. We also had clues that these two forms of therapy share some mechanisms of action. The challenge for us was to figure out a rational or scientifically based regimen. This led us to show that starting anti-PD-1/L1 therapy briefly before adding MAPK-targeted therapy was most rational in extracting durable tumor control or elimination, and in augmenting these shared mechanisms.
GM: Although effective drugs exist to help treat melanoma, resistance to therapy almost always arises. We have studied therapeutic resistance for the past 12 years, so the next step for us was to try to improve therapeutic approaches by a combinatorial/sequential strategy to avoid or forestall innate and acquired resistance.
What was the most surprising finding of this study for you?
RL: It was the finding that this sequential-combinatorial regimen could work so potently against melanoma brain metastasis. Melanoma is one of three or four cancers that most often metastasize to the brain in patients. Therapy resistance develops there more readily than in other organ sites. Metastasis to the brain also has an outsized, negative impact on patient survival.
GM: Our regimen was effective in different cancer models, even in melanoma brain metastases. This specific sequence AND combination of treatment could be an applicable therapeutic strategy for diverse cancers driven by MAPK pathway hyper-activation, ranging from melanoma to colorectal carcinoma to pancreatic adenocarcinoma, in both primary and advanced metastatic settings.
What was the coolest thing you’ve learned (about) recently outside of work?
RL: Usually, I run into cool things when I travel. Recently I have not traveled and, like many others, have spent more time with family. It was cool to realize that spending more time with my kids keeps me younger and not necessarily older.
GM: I recently visited the Louvre in Paris, France and discovered the behind-the-scenes care of this incredible museum. Over 2,000 workers take care of every piece of art – some are dedicated to removing the dust with a special pencil and vacuum, while others are renovating and restoring pieces using history-preservation techniques. More than 50 firefighters are also present on site to act quickly if a fire ignites or if the Seine river threatens to flood the museum. It was inspiring to learn about the Louvre’s commitment to preserving and sharing the legacy of humankind.



