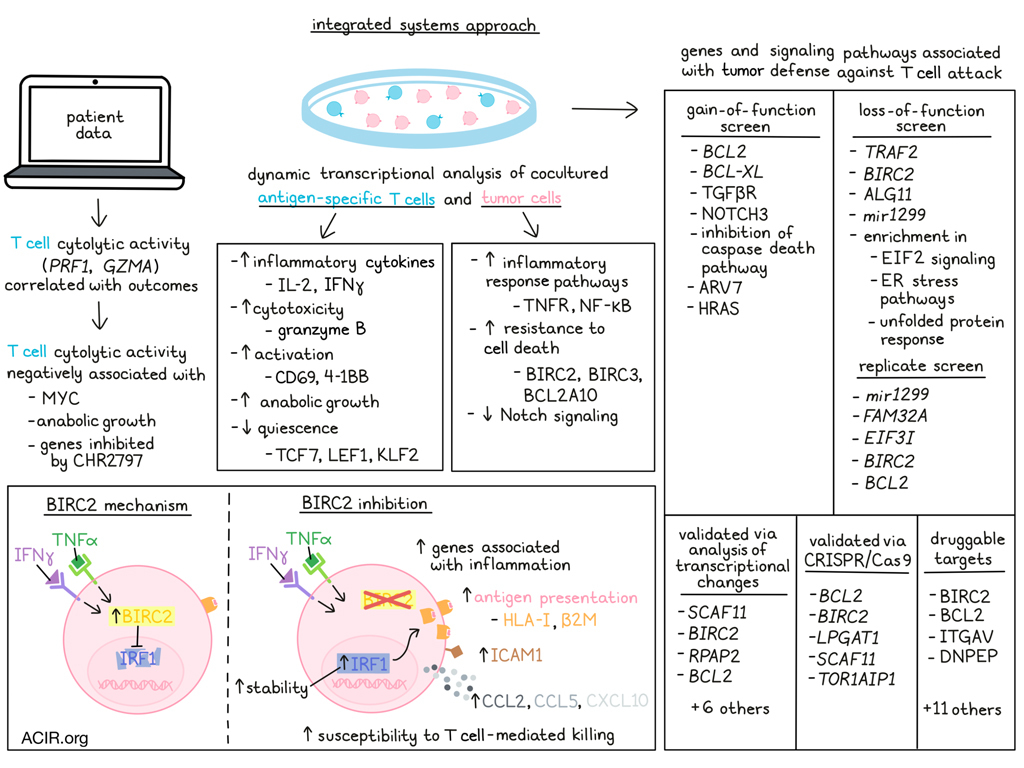
In cancer immunotherapy, research is often focused on improving the immune attack against cancer. However, Kishton and Patel et al. recently turned their attention towards how cancer defends itself against immune attacks, and how these defense mechanisms could potentially be targeted to enhance immune-mediated killing of tumor cells. To investigate tumor defenses, the researchers used an integrated systems approach to identify and interrogate tumor genes associated with resistance to cytotoxic immune activity. Their results were published in Cell Reports.
To begin, Kishton and Patel et al. evaluated data from patients with melanoma who were treated with ipilimumab, and found that while T cell abundance (as measured by expression of CD3E) was not significantly correlated with patient outcomes, T cell cytolytic activity, (as measured by expression of PRF1 and GZMA) was. In The Cancer Genome Atlas (TCGA) data from melanoma that was highly infiltrated by T cells, transcription-wide correlation analysis identified a number of genes that were negatively associated with cytotoxic activity, including MYC. Pathway analysis confirmed enrichment for MYC signaling, along with enrichment for anabolic growth and genes inhibited by CHR2797 (an aminopeptidase inhibitor) as negatively associated with cytotoxic activity, as well as with overall survival.
Next, the researchers used dynamic transcriptional analysis to study the kinetics of tumor gene expression upon interaction with T cells. In NY-ESO-1-specific human T cells co-cultured with NY-ESO-1-expressing Mel625 cells, mRNA expression was indicative of increased T cell production of inflammatory cytokines (IL-2 and IFNγ) and cytotoxic molecules (granzyme B), increased activation (CD69 and 4-1BB), and decreased quiescence (TCF7, LEF1, and KLF2). Pathway analysis further revealed upregulation of anabolic growth pathways in tumor-engaged T cells. In the Mel625 cells, expression of genes associated with canonical inflammatory response pathways, including TNFR and NF-κB pathways, were increased, along with genes associated with resistance to cell death (BIRC2, BIRC3, and BCL2A10). Expression of genes associated with Notch signaling was decreased in tumor cells after T cell engagement.
Utilizing functional genomics tools, Kishton and Patel et al. conducted a systemic gain-of-function screen using lentiviral transduction of constitutive-activating constructs to selectively activate pro-growth and -survival cell signaling pathways in A375 melanoma cells, which express NY-ESO-1. Constitutive activation of anti-apoptosis genes, BLC2 or BCL-XL, resulted in increased tumor cell resistance, as did constitutive activation of TGFβR or NOTCH3. Constitutive NOTCH1 activation, on the other hand, decreased PD-L1 expression, increased the β2M:PD-L1 ratio (favoring cytotoxicity), and sensitized tumors to cellular immunotherapy. Additionally, inhibition of the caspase death pathway was found to reduce β2M, and activation of AR-V7 or HRAS increased PD-L1; each of these alterations reduced the β2M:PD-L1 ratio, favoring tumor resistance. Similar results were observed in co-culture models of MART1-specific T cells and MART-1-expressing Mel62 cells.
Next, the researchers conducted a genome-scale loss-of-function screen using CRISPR/Cas9. In a co-culture of T cells and tumors in which 25% of tumors were eliminated within six hours (weak selective pressure), the researchers evaluated the abundance of single-guide RNAs, and identified TRAF2, BIRC2, ALG11, and mir1299 as tumor genes that likely disfavored T cell-mediated killing of tumor cells. Enrichment in EIF2 signaling, ER stress pathways, and the unfolded protein response were also identified in tumor resistance. A replicate screen identified mir1299, FAM32A, EIF3I, BIRC2, and BCL2.
To validate the gene identified in the screen as being associated with tumor defense, the researchers assessed T cell-induced transcriptional changes in each gene. This identified 10 genes with significantly increased expression after T cell engagement, including SCAF11, BIRC2, RPAP2, and BCL2. Targeting each of these genes with CRISPR/Cas9 validated the functional roles of 5 genes – BCL2, BIRC2, LPGAT1, SCAF11, and TOR1AIP1 – as knockout of each of these genes increased tumor cell death upon co-culture with tumor-specific T cells.
Having identified a number of genes associated with tumor defense against T cell attack, the researchers assessed the potential for tearing these defenses down to enhance immunotherapy. To this end, the team utilized a pipeline (Drug Gene Interaction Database; DGID) to identify potential targets for small molecule or biologic inhibition. This identified 15 targets for which inhibitory drugs were available, including BIRC2, BCL2, ITGAV, and DNPEP. Assessing the impacts of the inhibitory drugs on co-cultures of NY-ESO-1 T cells and A375 melanoma cells, the researchers found that several inhibitors enhanced T cell-mediated killing of tumor cells. Similar effects were also observed in co-cultures that included different target antigens (including neoantigens) and cancer types.
Taking a closer look into one particular target, BIRC2, the researchers investigated the mechanism by which it contributes to tumor defenses, and found that tumor cells increased BIRC2 expression upon exposure to T cell cytokines IFNγ or TNFα. Pharmacological inhibition or genetic ablation of BIRC2 in tumor cells increased T cell production of IFNγ and TNFα, indicating enhanced recognition of tumors. When BIRC2 was inhibited or knocked out, tumor cells increased expression of genes associated with inflammation, inflammatory signaling pathways, inflammatory cytokines, immune regulators, and antigen presentation pathways. Consistent with this, the researchers noted upregulation of the transcription factor IRF1 and IRF1-related transcriptional activity; increased surface expression of HLA-I and β2M; and increased total expression of HLA-1; and increased ICAM1. The increase in IRF1 was found to be due to increased IRF1 stability in the absence of BIRC2, and was also found to be essential for the increase in tumor susceptibility to T cell killing. These results suggest that BIRC2 limits IRF1-mediated upregulation of antigen presentation pathway genes, thus reducing T cell recognition of tumor antigens. Additionally, BIRC2 inhibition increased tumor cell production of CCL2, CCL5 CXCL10, and increased T cell migration towards tumor targets, in line with previous studies.
Finally, investigating BIRC2 targeting in vivo, the researchers switched to a murine system and found that pharmacological inhibition of BIRC2 improved the ability of adoptively transferred pmel T cells to control B16 tumor growth and extend overall survival, with the majority of mice achieving complete regressions. Genetic ablation of BIRC2 in tumor cells had similar effects, suggesting that targeting BIRC2 has the potential to enhance immunotherapy.
In summary, Kishton and Patel et al. used a systems approach to identify a number of genes and pathways associated with tumor defense against T cell-mediated attack. This allowed for the identification of several molecules, including BIRC2, that can potentially be targeted to reduce tumor cell defenses and enhance responses to immunotherapy in the future.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first co-authors Rigel Kishton and Shashank Patel, along with lead author Nick Restifo, answered our questions.

What was the most surprising finding of this study for you?
We were surprised to find how quickly tumor cells can act to defend themselves in the face of an aggressive T cell attack. These defensive genes are being upregulated within just a few hours of T cell engagement with the tumor, and they clearly have a functional role to help the tumor stay alive in this situation. It was also surprising how profoundly it can impact the ability of T cells to kill tumors when you utilize small molecules or biologics to target these tumor defense mechanisms in conjunction with the tumor-specific cells. It really gives a big boost to the therapeutic effect!
What is the outlook?
The notion of coupling existing small molecules or biologics with cell therapy in the clinic is really intriguing. If you can get these increases in therapeutic efficacy that we’ve seen in preclinical models, it may be clinically meaningful. Although that sort of strategy obviously creates clinical and logistical challenges – cell therapy is already pretty taxing on the patient and clinical infrastructure, with lymphodepletion prior to cell infusion, and sometimes giving IL-2 afterwards. Adding in another small molecule to the platform will take some work. This might be a little easier with some of the compounds we saw efficacy with, like birinapant and tosedostat, because they’ve been in the clinic in other settings.
What is the coolest thing you’ve learned recently outside of work?
Each of us has ventured out into the industry of science. As is clear from our scientific efforts, each of us seeks to fully turn novel scientific insights into treatments for patients. Each of us continues to love to deeply reveal biologic principals – especially questions underlying cancer immunotherapies – but we want to use this information not only to get scientific promotions, funding, and awards, but to directly help those afflicted by cancer. Thus, the science is the same and the work carries many of the same skills and passions, but the scale of the work, and the style of communication are very different between industry and academia. It seems clear that the right mix of industry and academia is the most efficient path to breakthrough therapies.



