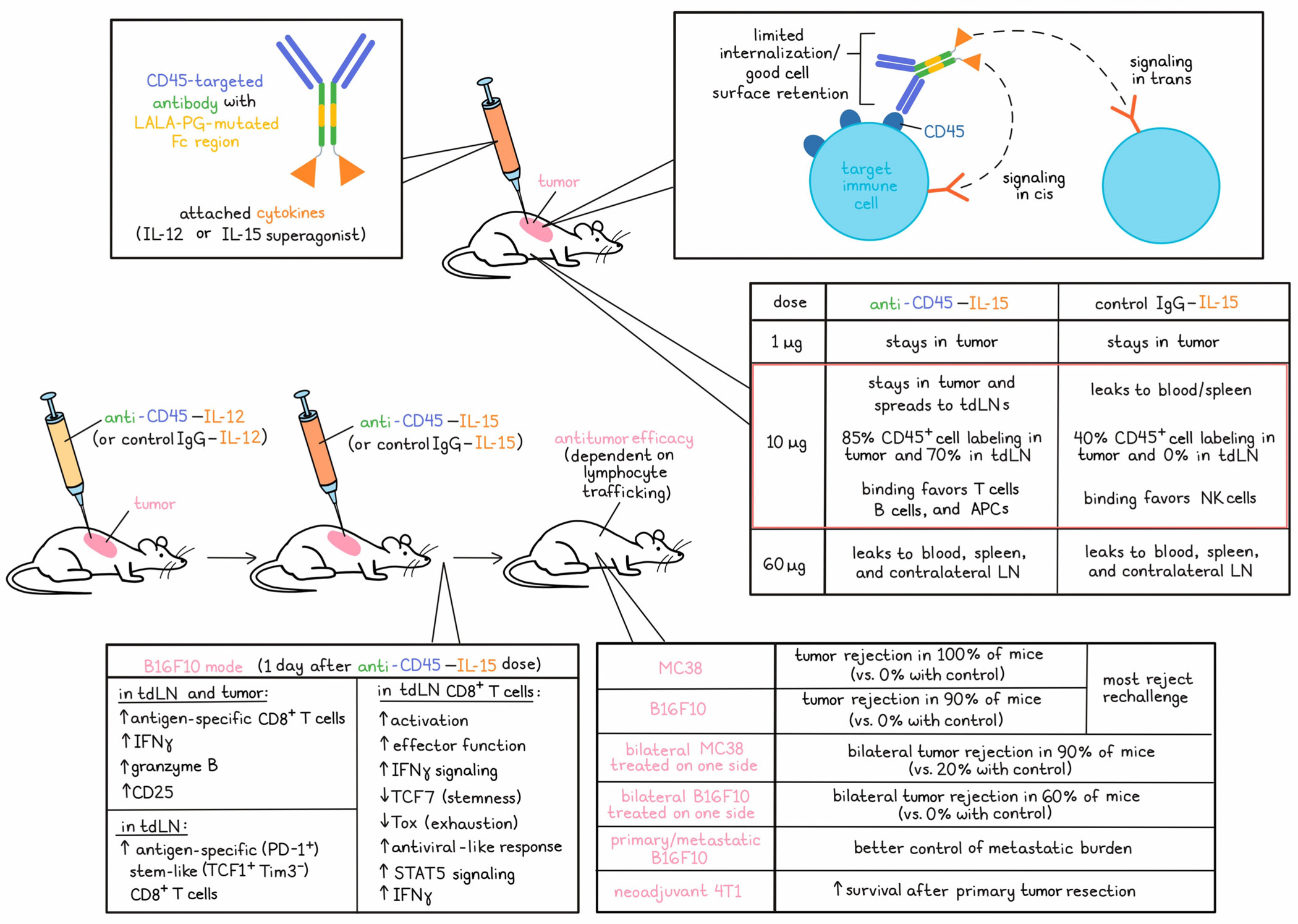
Cytokine therapy holds great promise in the oncology field, yet its application has been hindered by serious on-target, off-tumor effects. As a result, engineering approaches to confine cytokine activity to the tumor site have garnered considerable interest. Although advances in delivery methods have opened the intratumoral (i.t.) route as a viable option, rapid cytokine leakage from the injection site remains a critical barrier to both efficacy and safety. In a recent Nature Immunology paper, Santollani et al. devised an i.t. delivery strategy to selectively induce on-target cytokine activity in the tumor and tdLN to enhance systemic immunity while minimizing peripheral exposure.
Previous approaches from the Wittrup and Irvine labs to promote i.t. retention have broadened cytokines’ therapeutic window to promote primary tumor regression at safe doses, but have not yielded significant effects upon distal lesions, which substantially limits their practical applications. To overcome this problem, Santollani et al. engineered anti-CD45–cytokine fusions to selectively target lymphocytes upon i.t. Injection, as their prior work had shown that CD45 was expressed at very high levels on immune cells and was poorly internalized, which would allow for prolonged surface retention of the tethered cytokine. Past studies have also shown that a dose-optimized IL-12/15 regimen can lead to superior priming of T cells in both the tumor and tdLN, and to systemic immunity against distal untreated lesions in multiple mouse models, supporting IL-12 and IL-15 as useful payloads to investigate.
To engineer CD45-targeted cytokines, the team fused either IL-12 or an IL-15 superagonist to the heavy chain of an anti-CD45-IgG2c, which carried LALA-PG mutations to block Fc receptor signaling and thereby prevent cytotoxicity against cytokine-carrying cells. In all studies, targeted agents were compared to cytokines fused to a size-matched nonspecific IgG. CD45-targeted fusions maintained cytokine bioactivity both before and after binding to CD45, and exhibited a cell surface retention half-life of 24 hours on CD8+ T cells in vitro, compared to <1 hour for untargeted cytokines. Because this property could enable stimulation of neighboring cells, signaling capacity in trans and in cis were investigated and both were found to play important roles in the stimulation of responses. Similar results were observed across murine and human T cells, as well as with the cytokine IL-12, demonstrating the versatility of the approach.
To understand the effects of CD45 targeting on tumoral retention, biodistribution in mice was measured within a wide range of doses from 1 to 60 µg. Tumor retention was highly dose-dependent, which is unsurprising, given the dependence of site retention on the relative amount of accessible CD45 molecules in the tumor. At a dose of 1 µg, both CD45-targeted and control IgG–cytokines were confined to the tumor. At 10 µg, CD45-targeted cytokines disseminated only to the lymph nodes, while control cytokines showed leakage into the blood and spleen. The high 60 µg doses led to systemic accumulation of both CD45-targeted and control cytokines, although peripheral levels were higher in the control group. CD45-targeted cytokines at the 10 µg dose level promoted near-complete labeling of intratumoral CD45+ cells at ~85%, compared to ~40% in the control group, and robust ~70% labeling in the LNs compared to no significant labeling by the control. Importantly, minimal CD45-targeted cytokine labeling was observed in the periphery at this dose level. Labeled subsets also differed between the two groups; while control cytokines demonstrated a preferential affinity for NK cells, CD45-targeted cytokines bound to T cells, B cells, and DCs.
To assess antitumor efficacy, the researchers utilized a regimen in which an initiating dose of IL-12 was used as a priming agent to induce inflammation, in situ vaccination, and new T cell infiltration, followed by a dose of IL-15 several days later to amplify the newly primed response. In mouse models of MC38 or B16F10, CD45-targeted cytokine therapy led to 100% and 90% rejection rates, respectively, while no IgG control cytokine-treated tumors were cured. Most (14/18) mice rejected a rechallenge, suggesting induction of immune memory, which was consistent with the critical role of cDC1s and CD8+ T cells assessed through depletion studies.This effect was also dependent on both IL-12 and IL-15, and on the i.t. Injection route.
The critical goal of this study was to elucidate a localized cytokine therapy regimen that could also control distal tumors, including metastases. Accordingly, efficacy was assessed in a bilateral MC38 flank model, with cytokine injection into the right flank tumor only. 90% of CD45-targeted treatment mice rejected both tumors, while only 20% of control cytokine-treated mice did the same. In the more aggressive B16F10 model, 60% of CD45-targeted mice were cured, while all control mice succumbed to outgrowth of distal lesions. This effect was dependent on lymphocyte trafficking, as blocking egress from lymphoid tissues eliminated efficacy. Anti-CD45 cytokines also demonstrated superior efficacy in a primary/metastatic B16F10 model, as assessed by metastatic burden, and in a neoadjuvant 4T1 model, as assessed by mouse survival after primary tumor resection.
Localization of CD45-targeted cytokine therapy to the tdLNs motivated further assessment of the phenotypic changes leading to distal tumor control. Tumor antigen-specific CD8+ T cells in the tdLNs from mice bearing B16F10 melanoma profiled 1 day after anti-CD45–IL-15 treatment as part of the anti-CD45–IL-12/15 combination regimen exhibited genes associated with effector function and IFN signaling, and downregulated transcriptional regulators of stemness (Tcf7) and exhaustion (Tox). These cells were enriched in transcripts associated with antiviral responses, while cells from untreated tumors possessed a more canonical tumor response-like signature. Principal component analysis showed that following treatment with CD45-targeted and control cytokines, antigen-specific T cells clustered differentially, with gene set enrichment for STAT5 signaling and differential expression of IFNγ and other effector genes.
These results were affirmed experimentally by examining STAT5 phosphorylation and IFNγ production by antigen-specific T cells following treatment in the B16F10 model. Only CD45-targeted treatment promoted expansion of antigen-specific CD8+ cells in the TLDNs, as well as enhanced expression of IFNγ, Granzyme B, and CD25. Similar results were observed in the tumor microenvironment. The source of this increased population of effectors was likely antigen-specific (PD-1+) stem-like (TCF1+Tim3-) T cells in the tdLNs, which were enhanced ~6-fold by CD45-targeted therapy, although the proportion of these cells among antigen-specific CD8+ cells was decreased, likely due to effector differentiation.
In total, these results suggest that CD45 anchoring promotes on-target cytokine activity in the tumor and tdLN, and that lymphocyte tracking to distal sites is a critical mediator of the observed systemic immune effects in mouse models. Promising areas of future research include targeting cytokines to other immune cell receptors such as PD-1, along with developing rational combinations with other agents.
Write-up by Morgan Janes, image by Lauren Hitchings
Meet the researcher
This week, first author Luciano Santollani and senior co-authors Darrell J. Irvine and K. Dane Wittrup answered our questions.

What was the most surprising finding of this study for you?
The vigor of the abscopal response from such a minimalist dosing schedule was the biggest surprise for us. It demonstrates that T cell phenotypes that overcome the immunosuppressive mechanisms of distal untreated tumors are attainable even in the absence of supportive systemic checkpoint blockade therapy – in these model systems, at least.
What is the outlook?
The outlook will depend on all the uncertainties of any new biotech venture, but we intend to find a path to a clinical trial for this approach.
What was the coolest thing you’ve learned (about) recently outside of work?
KDW: The CH/Pi interaction between tryptophan and proline can stabilize a cis-amide bond at the proline. (Probably doesn’t sound so cool out of context; will have to wait to hear the whole story sometime in the future.)
DJI: My son is starting graduate studies in marine biology at the University of Hawaii, and one of his hobbies if photographing tiny sea life (think millimeter-scale). I expected Hawaii to have a vibrant biodiversity, but I am learning how really incredible that diversity is from his instagram posts!
LS: I recently came across a paper (link here) on mathematical modeling of espresso extraction. As a coffee nerd and engineer, I never thought I'd use the Navier Stokes equations to help me pull the perfect shot.



