T cell receptors (TCRs) are highly specific for peptide–MHC (pMHC) complexes, allowing for recognition of specific intracellular antigens, and making them extremely useful in targeting specific target cells. However, TCRs that target specific pMHCs are often difficult to identify. In lieu of identifying TCRs, Liu, Greenwood, Bonzanini, et al. set out to identify and generate small, stable proteins that recognize specific MHC-bound peptides (pMHCIs) of interest, thereby allowing for cellular targeting akin to TCR recognition. Their results were recently published in Science.
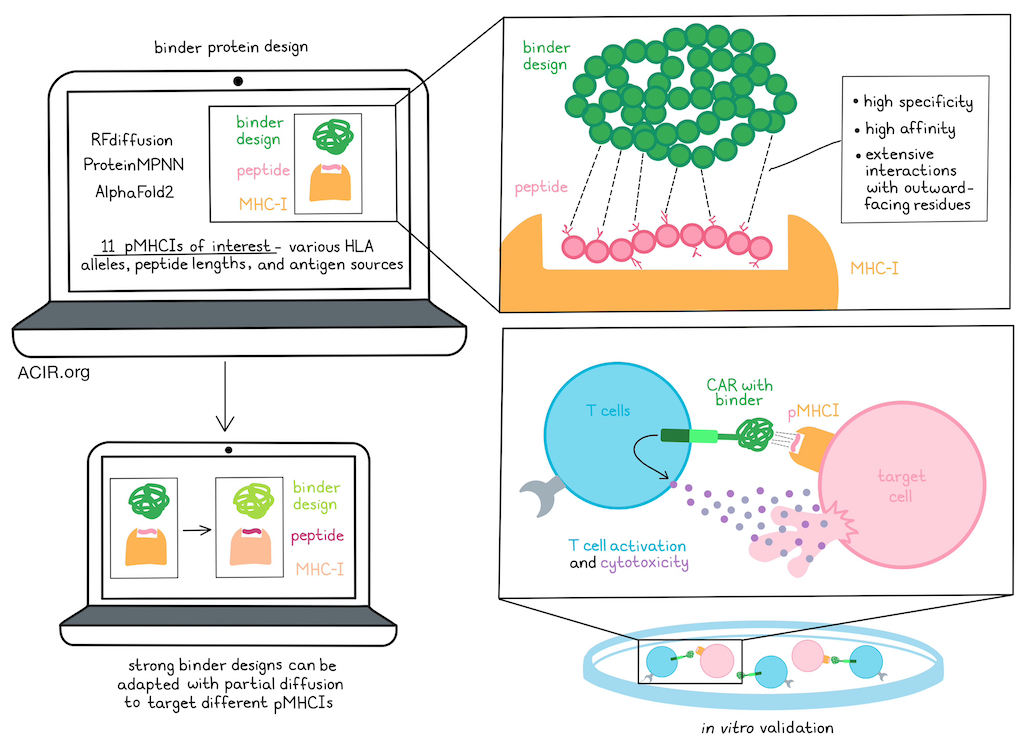
To computationally design high-affinity and high-specificity binders for peptides binding to class I MHCs (pMHCIs), Liu, Greenwood, Bonzanini, et al. utilized the generative AI method RFdiffusion to design proteins that would make extensive contacts with the outward facing residues of peptides presented in the MHC groove. ProteinMPNN and AlphaFold2 were also utilized to optimize protein sequences for folding and binding. Designs that were predicted to bind to on-target peptides considerably more than off-target peptides would then be selected.
Putting their design pipeline to the test, the researchers selected 11 target pMHCIs of interest, representing a diversity in HLA alleles (A*01:01, A*02:01, A*03:01, and C*07:02), peptide lengths (9-mers and 10-mers), and antigen sources (viral antigens, tumor-associated antigens, and neoantigens). For each pHMC-I target, the researchers obtained oligonucleotide pools encoding 200-12,000 designs, displayed them on yeast, and selected designs that specifically recognized the targeted pMHCI, but not closely related off-target peptides on the same MHC. For 8 of the pMHCI complexes, the researchers used RFdiffusion to generate de novo designs with a range of topologies and peptide interfaces that bound the target peptide. To evaluate the accuracy of their designs, they used a crystal structure of one of their binders – mart1-3, specific for MART-1 pMHCI – and found that it closely matched the design model at both the backbone and side-chain levels, and recapitulated the design’s interactions with the peptide.
Next, the researchers tested the functionality of their binders by either displaying them on yeast or incorporating them into CARs expressed on Jurkat cells. This allowed the researchers to identify designs that were specific for each of 3 pMHCI targets derived from viral peptides and presented on A*02:01, as well as for each of 4 pMHCI targets derived from tumor-associated antigens. The extensive contacts between the binders and their target peptides that had been predicted in the design models could also be validated experimentally.
While this strategy of using de novo RFdiffusion effectively generated binders to pMHCI targets, it was computationally expensive, as only a small fraction of trajectories yielded backbones that interacted extensively with the target peptide, without binding to the MHC molecule or off-target peptides. To overcome this, the researchers hypothesized that they could repurpose strong design backbones that effectively targeted one pMHCI to target others. To accomplish this, they used partial diffusion to adapt a strong binder design, mage513, specific for a melanoma-associated MAGE-A3 peptide presented on A*01:01, to additional 9-mer and 10-mer peptides derived from other tumor-associated antigens (gp100, MART-1, and PRAME) and presented on A*02:01. Using this system, the researchers were able to identify binders for each of the other peptides that were highly specific and made extensive interactions with the target peptides. When select designs were expressed as soluble proteins and purified, the researchers were able to use surface plasmon resonance to demonstrate that they bound to their cognate pMHCIs with high affinities.
To determine whether their novel binder designs could be utilized to confer T cell activation upon target recognition, Liu, Greenwood, Bonzanini, et al. incorporated the mage-513 design into CARs to be expressed on Jurkat cells. Upon incubation with 293T cells (which naturally express HLA-A*02:01 and C*07:02, and have an intact antigen presentation system) treated with MAGE-A3 or similar off-target peptides, the researchers found that cells expressing a CAR were strongly and specifically activated by MAGE-A3 peptide stimulation. Interactions between the design model and the predicted structures were consistent. Further, the researchers identified a particular residue, D29, for its role as a gatekeeper, likely by clashing with off-target peptides with bulky residues at a particular site. Other MAGE-A3-specific designs were also effectively stimulated by MAGE-A3 peptides, but showed weak or background activation.
To further characterize mage-513, the researchers used it to probe a library of peptide–HLA-A*01:01 complexes displayed on yeast, revealing top-hit peptides. Target cells pulsed with these top-hit peptides and incubated with mage-513 CAR-expressing Jurkat cells identified 3 activating peptides. An in silico scan of cross-activating peptides in the human proteome followed by testing for stimulation of mage-513 CARs revealed that the two most activating peptides shared similar outward facing features with those identified in the yeast display.
Next the researchers further evaluated CARs incorporating binders to gp100, MART-1, WT1, SARS, and HIV antigens, and found that these too could selectively activate Jurkat cells. Alanine scanning experiments showed that substituting various residues reduced this activation.
Given that many potential targets, like the PRAME protein, do not have experimentally determined structures, the researchers investigated whether they could design binders for such predicted pMHCI structures. Indeed, using their system, they identified two binders that, when incorporated into CARs, induced highly specific Jurkat activation upon incubation with 293T cells pulsed with PRAME. The observed specificity was consistent with the design model. Similar results were observed for binders designed to target the PHOX2B peptide.
Finally, to determine whether specific activation could be converted to specific target cell killing, the researchers transduced primary human T cells with CARs incorporating designed binders specific for PRAME or WT1 peptides, and found that both designed binder CAR T cells induced specific killing of HLA-A*02:01+ target T2 cells pulsed with their respective target peptides.
These results demonstrate the use of new protein design technology to identify highly specific binders to pMHCI complexes, overcoming many of the challenges associated with identifying specific TCRs, and allowing for comparable recognition of pMHCI molecules. When incorporated into CARs, these binders had the capacity to activate T cells and induce specific target cell killing. In similar research published in the same issue of Science, Johansen, Wolff, Scapolo, et al. and Householder et al. utilized similar protein design strategies to develop binders to NY-ESO-1 and to a neoantigen of unknown structure. Together, these papers demonstrate a range of potential avenues for developing binders to pMHCs and effectively mimicking TCR functionality for use in immunotherapies.
Write-up and image by Lauren Hitchings



