Interferon is full of paradoxes. Some patients have cancers with a mutated IFN pathway, but still respond to immune checkpoint blockade (ICB). Other patients have high serum levels of IFNγ, but their cancer progresses with ICB treatment. IFNγ can boost immune function, but also promotes T cell exhaustion via upregulation of PD-L1. In a paper recently published in Cell, Benci and Johnson et al. set out to explore how the opposing effects of IFN affect the therapeutic response to ICB.
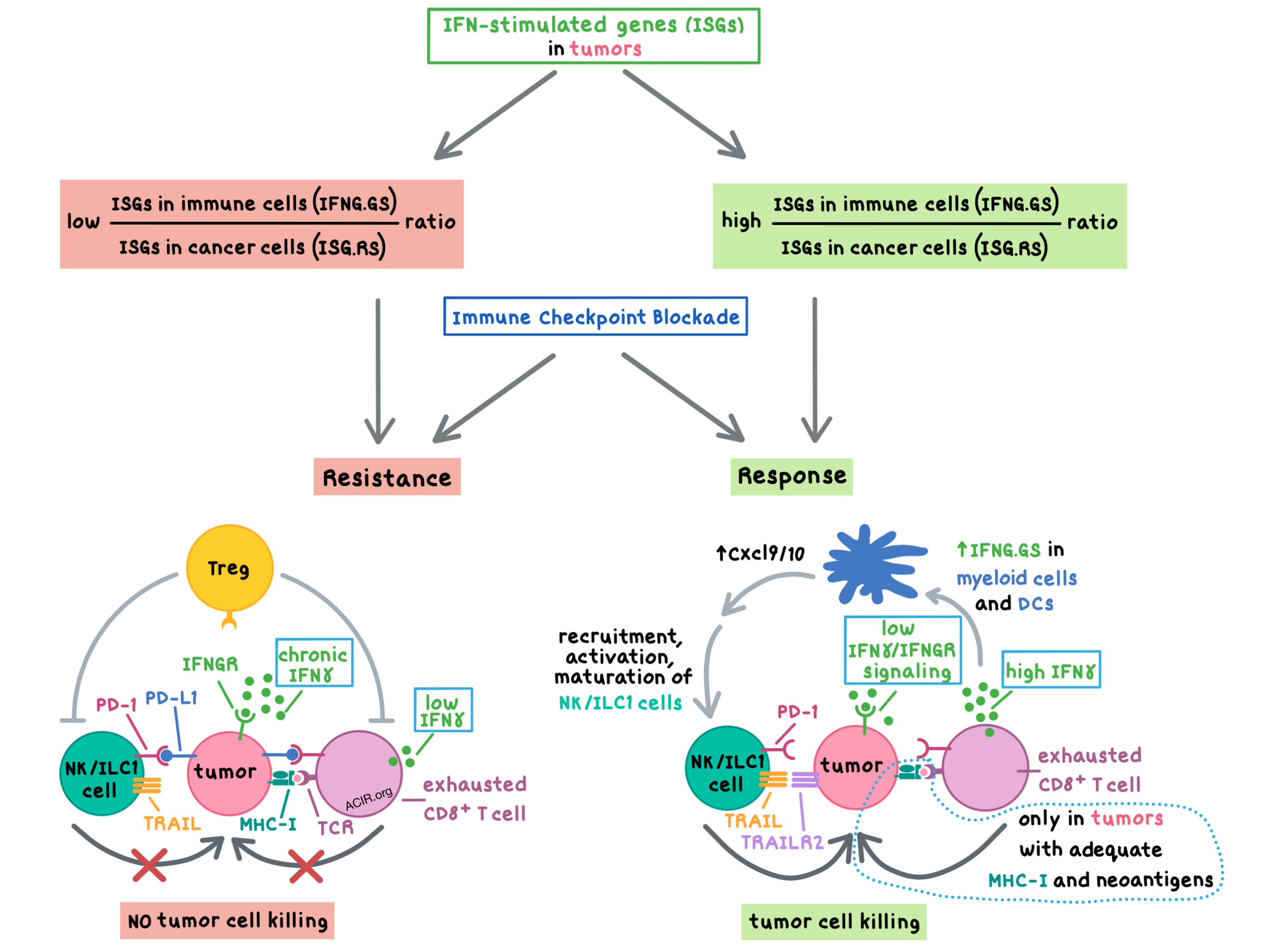
The researchers began by analyzing the single-cell RNAseq data of IFN-stimulated genes (ISGs) across various cell populations in human melanoma samples in TCGA. They found that the IFN-stimulated genes resistance signature (ISG.RS; initially associated with resistance to radiation and chemotherapy and later associated with resistance to immune checkpoint blockade) is mostly expressed by cancer cells, while a subset of the IFNγ hallmark gene signature chosen to not overlap with ISG.RS (IFNG.GS) is mostly expressed by the immune cells within the tumor, including T cells, NK cells, and macrophages. Low IFNG.GS/ISG.RS expression ratio correlated with resistance to anti-PD-1, while high IFNG.GS/ISG.RS ratio correlated with increased CD8+ T cells, activated NK cells, and response to anti-PD-1; this ratio predicted response independent of tumor mutational burden (TMB). These results indicated that IFN signaling in the tumor cells opposed the effects of IFN signaling in adaptive and innate immune cells to limit immune response.
In order to understand the mechanism behind the effect of the IFNG.GS/ISG.RS ratio, Benci and Johnson et al. utilized numerous tumor models that varied in baseline MHC-I expression, TMB, and neoantigen immunoediting, and prepared knockouts of either or both of the IFNα (type I) or IFNγ (type II) receptors in these tumors. They found that how the ratio of IFN signaling in immune cells (IFNG.GS) versus tumor cells (ISG.RS) affects various cell populations that participate in the antitumor response depends on context – particularly the MHC-I status and neoantigen availability (related to TMB) within the tumor.
In mice with CT26 colorectal tumors – which have high constitutive MHC-I expression and high TMB – knockout of type I and II IFN signaling in tumors slowed tumor growth and led to spontaneous regressions in some mice, and anti-PD-1 further boosted survival; complete responders were protected from rechallenge. The antitumor effect in this model was dependent on CD8+ T cells, which were able to be activated due to the maintenance of high levels of tumor MHC-I expression even in the absence of IFN signaling.
In contrast, Res 499 melanoma tumors, which are derived from B16F10 tumors, require IFN signaling for high MHC-I expression, and have a lower number of neoantigens than their parental tumors. In such tumors, knockout of the IFNγ receptor IFNGR improved response to anti-CTLA-4, and the antitumor response was dependent on NK1.1+ innate immune cells (including NK cells and innate lymphoid cells [ILC1s]), was independent of perforin, and did not protect against rechallenge. In this model, knocking out IFNGR in the tumors decreased tumor MHC-I expression and therefore, CD8+ T cells were not involved in direct killing of the tumors. Surprisingly, however, depletion of CD8+ (but not CD4+) T cells negatively affected IFNGR-knockout Res 499 tumors’ response to anti-CTLA-4, suggesting that CD8+ T cells play a supporting role in this tumor model.
Exploring the indirect role of CD8+ T cells in the IFNGR-knockout Res 499 tumor model, Benci and Johnson et al. found that IFNGR knockout in the tumor increased intratumoral terminally exhausted CD8+ T cells, and anti-CTLA-4 increased the CD8+ T cell production of IFNγ. The IFNγ in turn enhanced the IFNG.GS signature in myeloid cells and DCs, including the Cxcl9 and Cxcl10 genes that encode chemokines that help recruit, activate, and promote the maturation of NK cells. Furthermore, IFNGR knockout increased the proportion of TRAIL+ (TNF-Related Apoptosis Inducing Ligand) PD-1+ ILC1s. While IFNGR knockout in tumors decreased tumor PD-L1 expression, it increased the tumor expression of the TRAIL receptor TRAILR2. Decreased PD-L1 and increased TRAILR2 expression allowed for activation of NK/ILC1 cells, which mediated tumor killing in this model. Together, these results demonstrate that IFNγ produced by terminally exhausted CD8+ T cells is necessary for the NK/ILC1-dependent anti-CTLA-4 response in IFNGR-knockout Res 499 tumors. Interestingly, MHC-I expression on tumors was not required to activate CD8+ T cells, as cross-priming of tumor-specific T cells or activation of bystander T cells was sufficient to support NK/ILC1 function.
Having discovered the role of NK cells in the antitumor response to ICB, Benci and Johnson et al. once again looked at the human melanoma data and found that the proportion of activated NK cells inversely correlated with the proportion of Tregs in the tumor. In mice, the researchers found that Treg depletion played a role in the CT26 tumor model, and failure to deplete Tregs abrogated the response to anti-CTLA-4 in IFNGR-knockout Res 499 tumors. These observations suggest that Treg depletion may be required for optimal engagement of both innate and adaptive immune responses in the context of inhibited tumor IFNγ signaling.
Armed with the knowledge of the opposing mechanisms of IFN, the researchers analyzed human exome data of patients with non-small cell lung cancer (NSCLC) to explore whether mutations that decrease tumor IFN signaling may affect clinical response to ICB. They found that patients treated with anti-PD-1 plus anti-CTLA-4 who had tumor mutations that led to a decrease in ISG.RS gene expression experienced significantly longer progression-free survival compared to patients without such mutations. Consistent with preclinical data, tumors with such IFN variants had decreased PD-L1 expression, and the IFN pathway variants predicted response to ICB independent of TMB and PD-L1 expression.
Overall, Benci and Johnson et al. have begun to unravel the pro- and anti-tumor effects of IFN signaling by distinguishing signaling in immune cells (IFNG.GS) versus tumor cells (ISG.RS). Chronic IFN signaling in tumors coupled with immunosuppressive Treg cells leads to upregulation of PD-L1 on tumors, which contributes to the inability of exhausted T cells to directly kill tumor cells and to produce sufficient IFNγ to support NK/ILC1-driven tumor control. Immune checkpoint blockade with anti-PD-1 (stimulating exhausted effector T cells) or anti-CTLA-4 (depleting Tregs) unlocks the adaptive and innate immune cells, which can both contribute to tumor control dependent on the MHC-I status and neoantigen availability within the tumor.
by Anna Scherer
MEET THE RESEARCHER
This week, we asked first co-author Lexus Johnson a few questions about his research.
What prompted you to tackle this research question?
A number of groups have come to conflicting conclusions about how interferon affects the tumor microenvironment, so we were interested in figuring out how these opposite effects might fit together. Beyond that, we were curious how immune-edited tumors respond to immune checkpoint blockade despite a lack of T cell targets.
What was the most surprising finding of this study for you?
I think our most surprising finding in this paper is that NK cells can play such a large role in immune-edited solid tumors. Previously, they've received much more focus in liquid cancers, and it was exciting to find a major role for their function in this context.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently took a trip to Alaska and learned some new ocean fishing techniques. The biodiversity in the southeast part of the state is truly remarkable.



