Checkpoint blockade of the PD-1 axis (PD-1 or PD-L1) can be effective in inducing antitumor immunity, although many patients still relapse or fail to derive clinical benefit at all. Combinations of anti-PD-1 treatment with vaccines against tumor-associated antigens or with other immunomodulatory agents are actively being explored, and optimal timing and sequencing have yet to be established. In clinical trials combining a vaccine with anti-PD-1, anti-PD-1 treatments often begin earlier for logistical reasons. Recently, Verma et al. investigated whether the sequencing of anti-PD-1 and vaccine treatments might influence therapeutic outcomes; their results were published in Nature Immunology.
Investigating the common clinical sequence in which anti-PD-1 treatment is delivered early followed later by concomitant treatment with anti-PD-1 and vaccine, Verma et al. implemented this schedule in mice bearing TC-1 lung cancer or B16 melanoma (both resistant to anti-PD-1 and vaccines as monotherapies) and saw no significant therapeutic benefit. When no anti-PD-1 pre-treatment was given, however, coadministration of anti-PD-1 and vaccine induced a synergistic antitumor effect that slowed tumor growth. Profiling of T cell infiltrates from tumors showed that vaccine alone induced tumor antigen-specific T cells, and that this effect was even more significant when anti-PD-1 was coadministered with the vaccine. When anti-PD-1 pretreatment was administered, however, antigen-specific T cells decreased in response to subsequent vaccine or vaccine/anti-PD-1 combination therapy.
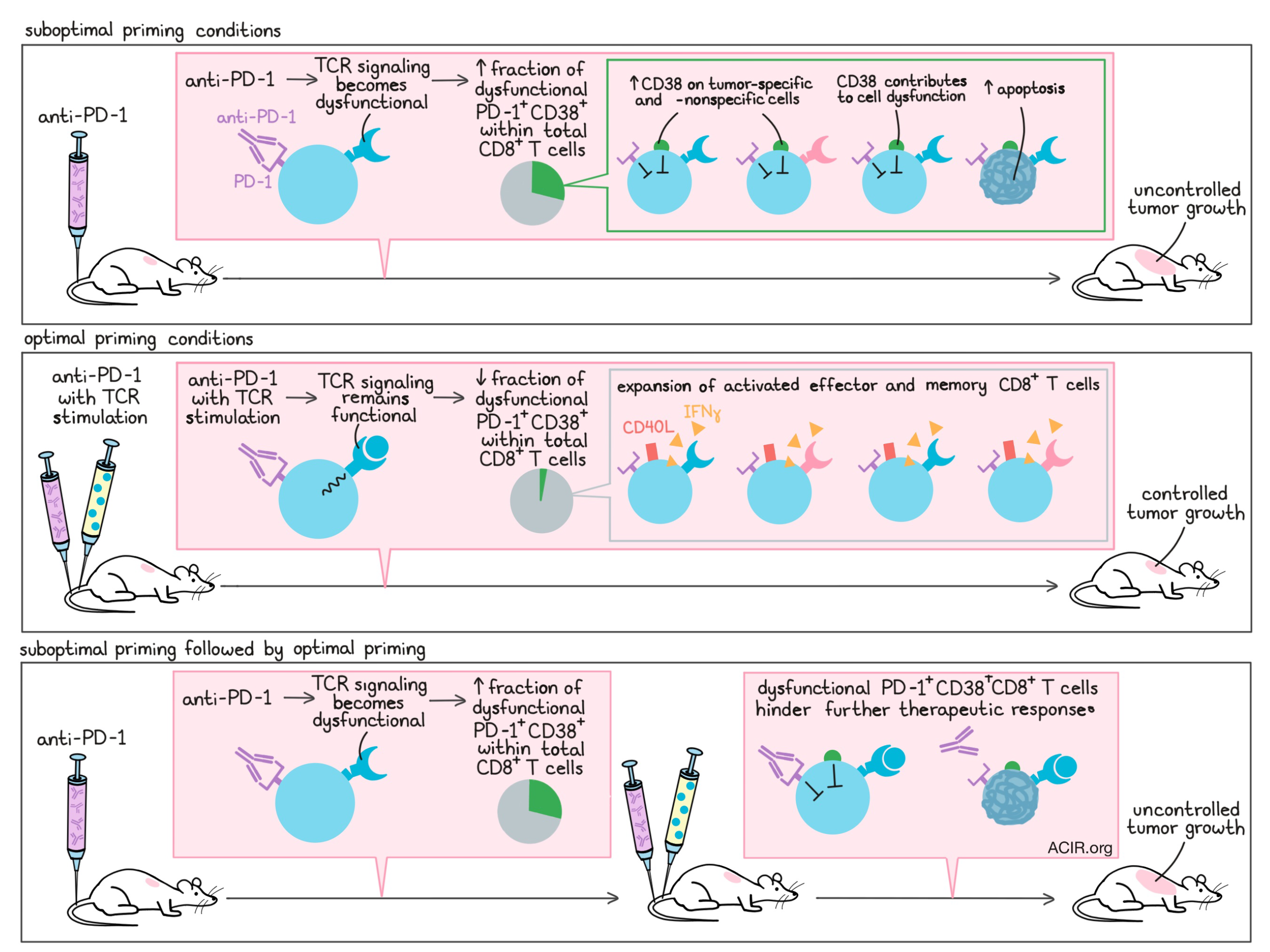
To better understand why anti-PD-1 pre-treatment hindered later therapies, Verma et al. examined overall and antigen-specific CD8+ T cell populations and identified signs of early and ongoing apoptosis in both. The researchers determined that anti-PD-1 did not enhance the activation of CD8+ T cells, and reasoned that apoptosis was not likely to have been caused by activation-induced cell death. Considering that anti-PD-1 could be causing cell death by altering the activation process, the researchers investigated the effects of anti-PD-1 and antigen stimulation in gp100-specific CD8+ T cells. When anti-PD-1 was administered in combination with gp100 peptide (to stimulate the TCR), TCR signaling was fully functional and cells took on an activated (CD40L+) effector (IFNγ-producing) phenotype. When anti-PD-1 was administered before gp100, however, it altered the phosphorylation patterns involved in TCR signaling, thereby interfering with TCR signaling in response to antigen stimulation. This drove CD8+ T cells towards a dysfunctional state, and ultimately towards their death.
PD-1+CD38hi CD8+ T cells are a subset of T cells that have been previously described as being dysfunctional and unresponsive to antigen restimulation, and Verma et al. found that pre-treatment with anti-PD-1 significantly increased overall CD38 expression, as well as the portion of PD-1+CD38+ T cells within both total CD8+ and antigen-specific CD8+ T cell populations. Vaccine treatment alone induced no change in CD38 expression, while coadministration of vaccine with anti-PD-1 decreased CD38 and the portion of PD-1+CD38+ cells within total CD8+ and antigen-specific CD8+ T cell populations. PD-1+CD38+ CD8+ T cells induced by anti-PD-1 pretreatment were dysfunctional (they did not show signs of activation upon antigen restimulation) and underwent apoptosis at an increased rate. Likely due to the increased rate of apoptosis within CD8+ T cells, mice pre-treated with anti-PD-1 also generated smaller fractions of central and effector memory CD8+ T cells.
To determine whether the PD-1+CD38+ CD8+ T cells that arose following anti-PD-1 pre-treatment were to blame for the failure of subsequent combination therapy, Verma et al. activated gp100-specific T cells in vitro in the presence of anti-PD-1 and adoptively transferred the total cultures or cultures depleted of PD-1+CD38+ cells (about 30% of the total culture) into mice. The researchers found that the PD-1+CD38+-depleted culture more effectively reduced tumor growth and increased survival. When expression of CD38 was genetically knocked down in cultures of dysfunctional PD-1+CD38+ CD8+ T cells, the cells regained their ability to proliferate, to become activated, and to express effector molecules, linking CD38 directly to dysfunctionality.
Given that anti-PD-1 pretreatment induced dysfunctional PD-1+CD38+ CD8+ T cells, while anti-PD-1 in combination with vaccine did not, Verma et al. investigated whether the quality of T cell priming may play a role. Using OVA-specific T cells in vitro, Verma et al. compared the impact of anti-PD-1 in the context of optimal priming (stimulation with high-affinity OVA) or suboptimal priming (no stimulation, or stimulation with low-affinity OVA-V). While blocking PD-1 under optimal priming conditions had no impact on PD-1+CD38+ CD8+ T cell production, blocking PD-1 under suboptimal priming conditions or before optimal priming drove T cells towards a PD-1+CD38+ phenotype and apoptosis-mediated cell death. Similar results were observed in tumor-bearing mouse models of suboptimal and optimal priming.
To determine the relevance of PD-1+CD38+ CD8+ T cells in human cancers, Verma et al. evaluated immune infiltrates from tumor biopsies from patients with metastatic melanoma taken before or after anti-PD-1 or combination anti-PD-1/anti-CTLA-4 therapy. They found that before and after checkpoint blockade, non-responding lesions contained a higher fraction of PD-1+CD38+ CD8+ T cells compared to responding lesions, suggesting that the fraction of PD-1+CD38+ cells within infiltrating CD8+ T cells in the tumor could serve as a biomarker of response. Further, most responders showed a decrease in the fraction of CD38+ cells within PD-1+CD8+ T cells at nine weeks post-therapy, while non-responders showed stabilization or an increase in CD38+ cells, which could serve as an easy-to-access biomarker of on-treatment response.
Overall, Verma et al. show that administration of PD-1 blockade under suboptimal priming conditions drives the production of a PD-1+CD38hi CD8+ T cell population that interferes with antitumor immunity and prevents therapeutic responses. This evidence may help to explain why many cancer patients fail to derive clinical benefit from checkpoint blockade and combinations, as well as why patients with a higher mutation burden (i.e. more opportunity for optimal on-site priming) are more likely to respond. Improving the antigenicity of tumors or correctly sequencing PD-1 blockade and vaccines in current and future clinical trials could serve as important strategies in improving antitumor responses.
by Lauren Hitchings
Meet the researcher
This week, lead author Samir Khleif answers our questions.

What prompted you to tackle this research question?
As a cancer immunologist and a physician who treats patients, it was wonderful to see anti-PD-1 therapy work for so many patients. However, at the same time it was painful to see many patients not getting the benefits out of their anti-PD-1 therapy. This had us thinking about the reasons that lead to the non-response in these patients. To address this issue, we turned our attention to model systems that could be employed to answer the question regarding resistance to anti-PD-1 therapy in humans. Accordingly, we made some observations where we saw that initial activation status of the killer lymphocytes had a great impact on the therapeutic outcomes seen after anti-PD-1 therapy. Steadily building upon our initial observations, we discovered this new mechanism of resistance to anti-PD-1 therapy wherein we found that a single dysfunctional cell type can be responsible for therapeutic failures.
What was the most surprising finding of this study for you?
The most surprising finding of this study was the observation that the very treatment agent by itself can turn rogue and contribute to failure of the therapy. Though, of course, other contributing factors, such as suboptimal priming conditions, should be there to make the environment conducive for generation of dysfunctional cells that render the treatment ineffective.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently visited the Amazon and, amongst the many things that I learned about the eco harmony and animal behavior is the beautiful synchronized musical symphony that cicadas make during the mating season. This was discovered by scientists using sound-slowing techniques. Besides the amazing musical tune that sounds like a Middle-Age Viennese Choir, it tells a lot about nature organization and lack of randomness in biology. It turned out that other species do the same, including crickets.



