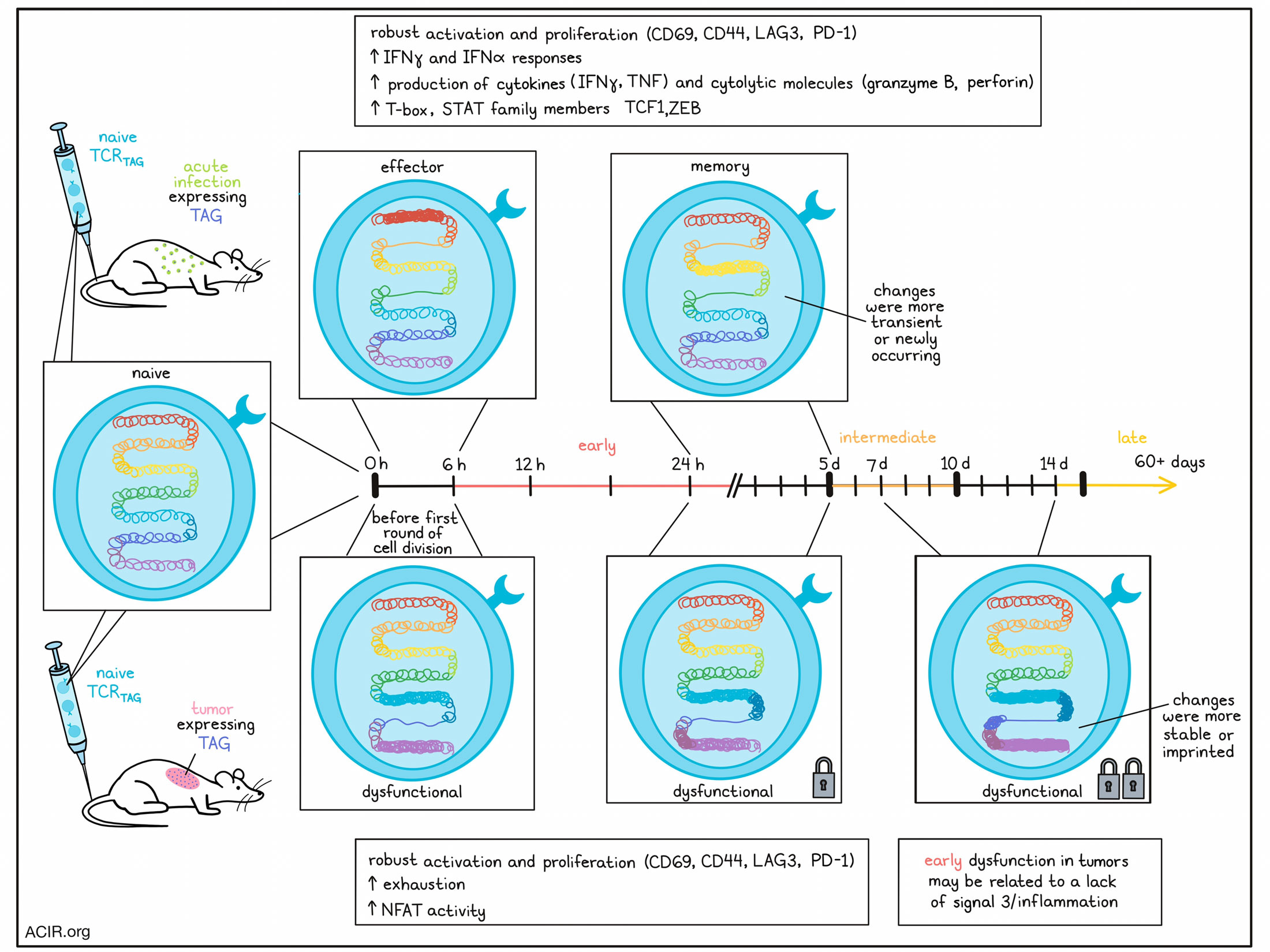
Tumor-specific CD8+ T cells can be strong effectors in the immune battle against cancer, but they are often limited by dysfunction/exhaustion induced by the tumor microenvironment. Rudloff et al. investigated this dysfunction by comparing differentiation, cell division, and chromatin accessibility in antigen-specific CD8+ T cells in tumor versus acute infection models. This revealed that dysfunction programming was initiated early in tumors, and was further “imprinted” with extended exposure to tumor/tumor antigens. Their results were recently published in Nature Immunology.
Having previously published results identifying features of tumor-induced dysfunction, Rudloff et al. utilized their established model of liver carcinogenesis to track tumor-specific CD8+ T cell division and differentiation over time. Naïve CD8+ T cells specific for TAG (SV40 large T antigen) were labeled with CFSE and transferred into mice with TAG-expressing tumors or TAG epitope-expressing Listeria infection. In both models, the TCRTAG-positive T cells (TCRTAG) robustly divided, expanded, and upregulated markers of activation, including CD69, CD44, LAG3, and PD-1. However, unlike cells from the acute infection model, cells from the tumor model failed to produce effector cytokines (IFNγ and TNF) or cytolytic molecules (granzyme B and perforin), and did not show evidence of degranulation (CD107a membrane localization) upon ex vivo restimulation. These differences were evident even at the 6-hour time point, before the first round of cell division. TOX was not yet induced at this time point, suggesting that it was not responsible for the impairment of effector functions.
To assess whether early dysfunction was associated with inadequate priming, the researchers tested whether committed effector T cells would become dysfunctional upon exposure to the tumor setting. When time-matched effector TCRTAG T cells from acute infection models were adoptively transferred into tumor models, they continued to proliferate, but began losing their capacity for cytokine production (TNF first, then IFNγ) within 12 hours, with complete loss of cytokine production in a week. For comparison, effector TCRTAG remained IFNγ/TNF double producers upon adoptive transfer back into the acute infection model, suggesting that tumor environments can override established effector programming. The results of these studies were recapitulated in a pulmonary metastasis model using B16-OVA and OT-I cells, suggesting that these findings were not specific to the tumor origin, tissue, or target antigen.
Next, Rudloff et al. turned their attention towards epigenetic remodeling. Using RNA and ATACseq, the researchers found that within 6 hours, TCRTAG from tumor and acute infection models had distinct chromatin accessibility and gene expression profiles, with the most changes occurring within the first 6 hours versus at 12 and 24 hours. Motif and gene set enrichment analysis showed that while TCRTAG from both models showed evidence of activation and TCR signaling relative to naive mice, those from acute infection models showed evidence of increased STAT family member activity and enrichment for IFNα and IFNγ responses. TCRTAG from tumor models, on the other hand, showed increased accessibility of chromatin associated with exhaustion. They also showed evidence of increased NFAT family member activity, consistent with expression of multiple inhibitory receptors and later expression of TOX, and enrichment for gene sets typically expressed in T cells stimulated in the absence of signal 3. These results suggest that while TCR stimulation was similar in both models, dysfunction in the tumor models may be associated with a lack of adequate inflammatory cytokine signaling during priming.
Gene set enrichment analysis of early TCRTAG (isolated within the first 6, 12, or 24 hrs), revealed that in acute infection, early TCRTAG were enriched for gene sets associated with late effector and memory phenotypes, while in tumor models, early TCRTAG were enriched in gene sets associated with late T cell dysfunction typically seen in tumors or chronic infection. Using their current and previously published data, the researchers found that chromatin accessibility in TCRTAG in tumors clustered into three groups based on the duration of tumor exposure: early (6–24 hours), intermediate (5–10 days) and late (14–60+ days). Here, the greatest number of changes in differentially accessible chromatin peaks occurred within 6 hours, with another large round of changes between 24 hours and 5 days, and a third smaller round between 7 and 14 days. However, nearly half of the day 5 chromatin accessibility profile was already established within the first 6 hours and remained consistent. In the infection model, only two rounds of major changes (within 6 hours and between 24 hours and 5 days) were observed in TCRTAG, and many of the changes were transient or newly occurring between 24 hours and 5 days, and only few changes were evident after day 5, suggesting that these cells likely established a memory-like state upon pathogen/antigen clearance.
MOtif aNAlysis with Lisa (monaLisa) identified NFAT enrichment as a key driver of transitional peak changes in tumors, consistent with mediating exhaustion, while T-box TF family motifs were drivers of peaks changes in acute infection, consistent with mediating effector functions and establishing memory populations. In both models, these changes were established within 6 hours and were reinforced between 24 hours and 5 days.
To test the extent to which tumor-induced changes were reversible, TCRTAG were isolated from tumors at various time points and “parked” in tumor-free mice. When the cells were isolated from tumors earlier, they were more likely to downregulate PD-1 and begin producing cytokines than when they were removed from tumors at later time points, suggesting “imprinting” of effector function loss with extended exposure to tumor/tumor antigens.
This imprinting was also reflected in chromatin accessibility data, which showed peaks in TCRTAG from tumors at earlier time points were more transient, but became more stable with longer tumor exposure. It also showed that pre-parking samples clustered separately from post-parking samples and antigen-experienced memory T cells. PD-1 imprinted quickly, while TOX imprinting was established later. KLF family motifs with described roles in T cell quiescence, functional restraint and fixed dysfunction were already predominant in post-parking day 5 TCRTAG, whereas TCF1 and ZEB family motifs (evident in memory populations) were lacking. NFAT TF enrichment, however, was readily reversed upon parking, suggesting that it was likely maintained by continuous TCR signaling. Together these results suggest that while removing dysfunctional TCRTAG from tumors at early and intermediate time points may reverse some chromatin remodeling and expression of inhibitory molecules, effector functions are unlikely to be recovered, as transcription factors that induce effector and memory functions are not expressed.
Overall, these results show that epigenetic programming for dysfunction is initiated within hours after antigen encounter and before the first cell division of antigen-specific CD8+ T cells in tumors, and is further imprinted with ongoing exposure to tumor antigen over days and weeks. While some of this dysfunction programming is reversible early on, effector function is unlikely to be recovered without intervention.
Write up and image by Lauren Hitchings
Meet the researcher
This week, lead author Mary Philip and first author Michael W. Rudloff answered our questions.

What was the most surprising finding of this study for you?
MP: Going into the study, I thought that CD8+ T cells activated in tumors would initially be functional, but get progressively dysfunctional with each cell division. I was really surprised when even before they had undergone their first cell division, tumor-specific T cells had lost nearly all their cytotoxic capacity.
MWR: I was struck by just how much epigenetic and transcriptional change could happen within hours after a T cell was activated. To see such a large-scale chromatin remodeling event take place so quickly was fascinating and challenged my understanding of how fast these biological processes can occur in vivo.
What is the outlook?
MP: Immune checkpoint blockade is analogous to taking off the brakes from T cells, but our work suggests that the tumor-specific T cells may not even be moving in the first place. We need new strategies to jumpstart T cells in tumors, for example, by providing exogenous innate immune signals or engineering T cells to provide their own inflammatory signaling.
MWR: Given how quickly even previously functional effector CD8+ T cells can lose cytotoxic capabilities, we may need to implement approaches that not only boost the numbers of cytotoxic T cells, but can extend the duration of antitumor immunity for each cell as they infiltrate and activate in tumors.
What was the coolest thing you’ve learned (about) recently outside of work?
MP: This summer I have been learning to play the upright double bass, which is cool because you not only hear the music, but really feel those beautiful low tones resonating through the whole instrument and coming up through the floor.
MWR: This summer our youngest kid started crawling and walking, so my wife and I are experiencing the fun and excitement of two very mobile toddlers.



