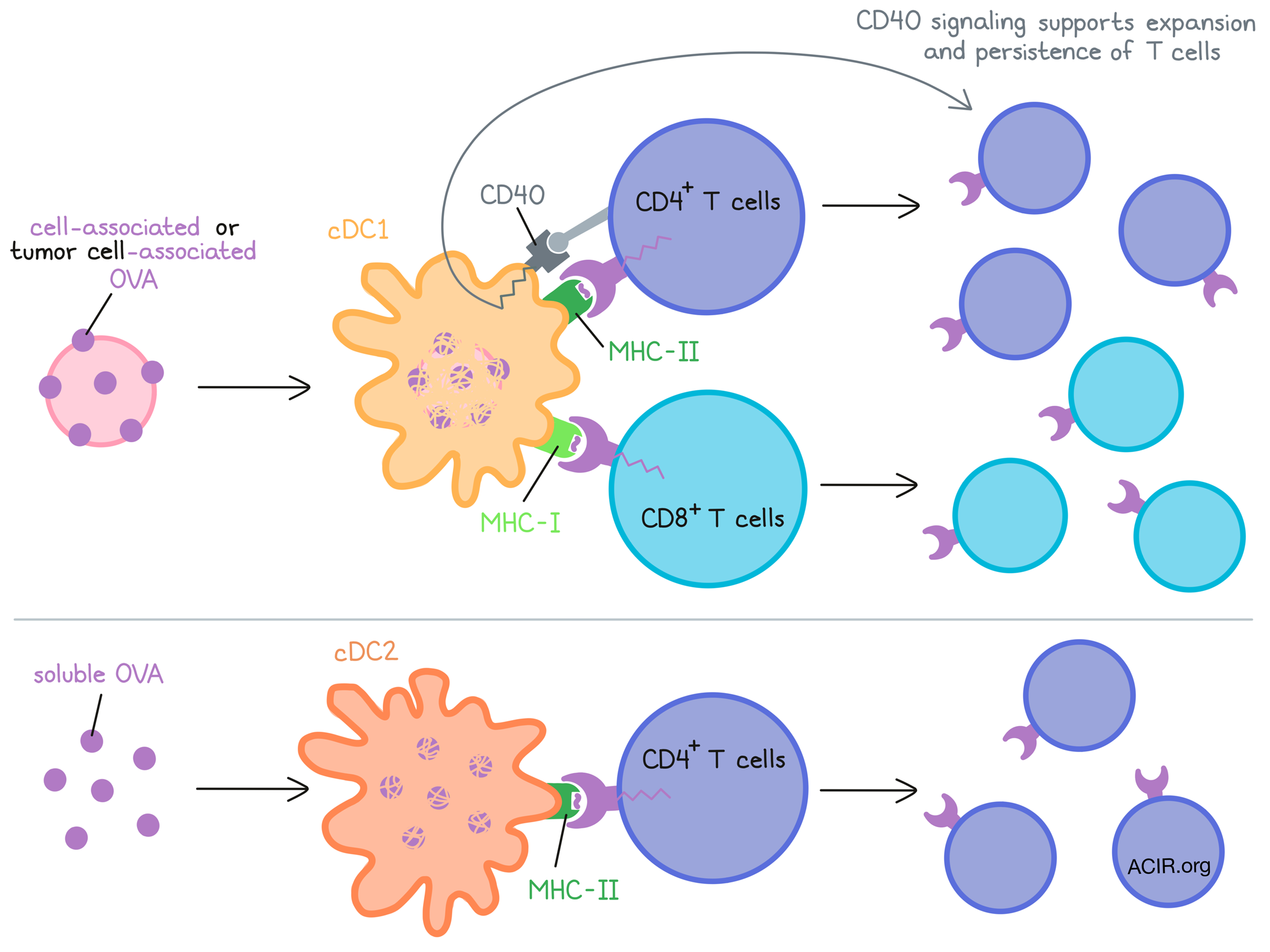
In the currently understood paradigm of T cell priming, it has generally been assumed that cDC1s cross-present peptides on MHC-I to prime CD8+ T cells, while cDC2s, which express high levels of MHC-II, present antigens on MHC-II to prime CD4+ T cells. CD4+ T cells are then thought to license cDC1s, supporting CD8+ T cells. However, the exact mechanisms of CD4+ T cell priming have not been shown in tumors, and some evidence has suggested that cDC1s might play more of a role than cDC2s in priming CD4+ T cells. Exploring the interplay between cDC1s, cDC2s, CD4+ T cells, and CD8+ T cells in a tumor rejection setting, Ferris, Durai, and Wu et al. uncovered evidence that challenges the current paradigm of CD4+ T cell priming; their results were recently published in Nature.
To study the cellular interactions that mediate tumor rejection, Ferris, Durai, and Wu et al. began by establishing the baseline immunologic features of a methylcholanthrene-induced progressor fibrosarcoma cell line (1956). 1956 was weakly immunogenic, and was not initially rejected by wild-type mice, but induced immunological memory following implantation and removal, protecting mice from rechallenge. When either CD4+ or CD8+ T cells were depleted in these mice, protection from rechallenge was lost, indicating that both T cell types were essential to the recall response. The researchers therefore posited that if both CD8+ T cells and CD4+ T cells were required, then priming via MHC-I and MHC-II must have also occurred, but which APCs were involved?
Some evidence in past studies has suggested that cDC2s are responsible for CD4+ T cell priming because of high MHC-II expression, while other studies have indicated that cDC1s may actually play this role. To study early priming, the researchers used a variant of 1956 that expressed either a membrane-associated ovalbumin (1956-mOVA) or an empty vector (1956-EV). The researchers first showed that 1956-mOVA could be cleared by the immune system without surgical resection in wild-type mice. When a mouse strain lacking DC1s (due to the absence of the transcription factor Irf8) was used, 1956-mOVA tumors were not cleared and an OVA-specific CD8+ T cell response was not detected.
Looking more closely at the role of CD4+ T cells, Ferris, Durai, and Wu et al. examined early CD4+ T cell proliferation in response to 1956-mOVA by transferring OT-II-transgenic CD4+ T cells, which are activated by interactions with MHC-II carrying an OVA peptide. In wild-type tumor-bearing mice, OT-II CD4+ T cells proliferated mainly in the draining lymph nodes, indicative of antigen recognition and activation. However, in mice lacking Irf8, and therefore cDC1s, much lower proliferation of OT-II cells occurred, suggesting that cDC1s played an important role in CD4+ T cell priming. Similar results were observed in B16F10-mOVA versus B16F10-EV melanoma models.
To determine whether the requirement for cDC1s in priming CD4+ T cells was due to cDC1s transporting antigens to draining lymph nodes and transferring them to cDC2s, the researchers investigated whether MHC-II presentation on cDC1s was essential to CD4+ T cell priming. When MHC-II was conditionally inactivated on cDC1s (using Xcr1cre mice), adoptively transferred OT-II CD4+ T cells proliferated in response to soluble antigens, but proliferation was reduced in response to cell-associated antigens (loaded osmotically into MHC-I- or MHC-II-deficient splenocytes followed by irradiation). Further, in a mouse model in which MHC-II was expressed exclusively on cDC1s, OT-II CD4+ T cell proliferation was still induced following exposure to cell-associated, but not soluble antigens. Together, these results suggested that MHC-II expression on cDC1s was necessary and sufficient to prime CD4+ T cell responses to cell-associated antigens, which are relevant in a tumor setting, while MHC-II expression on cDC2s could prime CD4+ T cell responses to soluble antigens, which are more relevant in other settings, like in allergic responses.
Next, the researchers wanted to confirm that this mechanism would hold true for tumor cell-associated antigens. In wild-type mice, which expressed both MHC-I and MHC-II on cDC1s, 1956-mOVA tumors were rejected. However, in mice lacking MHC-II expression on cDC1s, tumors were not rejected, suggesting that MHC-II expression on cDC1s was essential in tumor rejection. Interestingly, the expression of MHC-II by cDC1s affected optimal priming of both CD8+ and CD4+ T cells specific for the 1956-mOVA tumor, as mice lacking MHC-II on cDC1s showed significantly weaker expansion of endogenous OVA-specific CD8+ T cells. This indicated that the interaction between CD4+ T cells and cDC1 ultimately affected the antitumor CD8+ T cell response.
To better understand how the interaction between CD4+ T cells and MHC-II on cDC1s impacts CD8+ T cells, Ferris, Durai, and Wu et al. turned their attention towards CD40 signaling in cDC1s. In a mouse model in which CD40 was absent on cDC1s, but was expressed normally on cDC2s and B cells, mice failed to reject 1956-mOVA and showed greatly reduced expansion of endogenous OVA-specific CD8+ T cells. Similarly, these mice showed reduced proliferation of OT-I CD8+ T cells in response to immunization with cell-associated ovalbumin. This effect was not observed following transfer into CD40-deficient tumor-bearing mice, nor in cultures containing cDC1 from draining lymph nodes, suggesting that CD40 on cDC1s was not required for initial antigen presentation. However, the number of OT-I CD8+ T cells surviving past 3 days was reduced in mice with cDC1s lacking CD40, suggesting that CD40 signaling in cDC1s helps to support the expansion and/or persistence of antitumor CD8+ T cell responses. CD40 expression in cDC1s was also required to support the proliferation OT-II CD4+ T cells in response to cell-associated antigens, but not soluble antigens. Interestingly, the requirement for CD40 expression was not observed following inoculation with1956-mOVA tumors, which the authors attribute to differences in antigen burden in wild-type versus CD40-deficient mice due to tumor rejection in the former.
Finally, examining the location of cDC1s and CD40 expression, the researchers found that few cDC1s were present within tumors, and that CD40 expression in the tumor microenvironment was mainly attributed to B cells and macrophages. Instead, migratory cDC1s in lymph nodes were found to express CD40, suggesting that this was likely where CD40 signaling by cDC1s had its effect. Interestingly, the entry of CD4+ T cells into tumors was dependent on the presence of cDC1s in tumors, despite the fact that these cells were relatively rare in this location.
This new work by Ferris, Durai, and Wu et al. suggests that cDC1s are responsible for priming both CD4+ and CD8+ T cell responses to cell-associated antigens, including tumor cell-associated antigens, while cDC2s prime CD4+ T cell responses to soluble antigens. Further, CD4+ T cell interactions with MHC-II induce CD40 signaling within DCs, which further supports the expansion and persistence of antitumor CD4+ and CD8+ T cell responses, and ultimately the rejection of tumors. This research challenges and extends the current understanding of the mechanism of CD4+ T cell priming and its importance within tumors.
by Lauren Hitchings
Meet the researcher
This week, first author Stephen Ferris answered our questions.
What prompted you to tackle this research question?
Previous studies had shown that cDC1 do not present antigen to CD4+ T cells. However, CD4+ T cell "help" is required for rejection of many viruses and tumors, presumably through CD40:CD40L interactions (CD40 signaling in the cDC1 and CD40L provided by CD4+ T cells). If cDC1 do not present antigen to CD4+ T cells, this would mean that CD4 "help" would be non-cognate, i.e. non-specific. This would also mean that MHC-II expression on cDC1s is superfluous. We hypothesized that this is not the case and that in certain settings, cDC1s can present antigen to CD4+ T cells and receive the CD40L "help" simultaneously.
What was the most surprising finding of this study for you?
The most surprising finding was that not only was CD40 signaling within the cDC1 required for optimal CD8+ T cell priming, but also for optimal CD4+ T cell priming as well.
What was the coolest thing you’ve learned (about) recently outside of work?
Recently, the coolest thing that I have learned is that you can catch crayfish with a stick, string, and bread. My wife and our two kids, Wally (age 2) and Annie (age 4), have gone to our local pond and caught many crayfish over the past couple of months. It's been really fun to watch the kids learn how to catch them and learn about the anatomy of the crustaceans.



