While CD8+ T cells with various stemness, tissue residency, and exhaustion features have been implicated in antitumor responses and ICB efficacy, the exact contribution of each subset remains poorly understood. Wijesinghe, Rausch, et al. used multiple genetic murine models to investigate the functionality and relation to ICB efficacy of various T cell subsets in lymph nodes and in tumors. Their results were recently published in Nature Immunology.
The researchers used the transplantable AT3-OVA model, which contains high frequencies of TCF1+ and CD103+ intratumoral tissue resident (Trm)-like CD8+ T cells. Mice were treated with dual ICB, and tumoral CD44+CD8+ T cells and PD-1+CD8+ T cells from tumor-draining lymph nodes (TDLNs) were isolated and subjected to RNAseq and TCRseq. Clustering analysis revealed that clusters of precursors of exhausted T cells (Tpex) and terminally exhausted T cells (Tex) were largely exclusive, but Trm signatures overlapped with both Tpex and Tex.
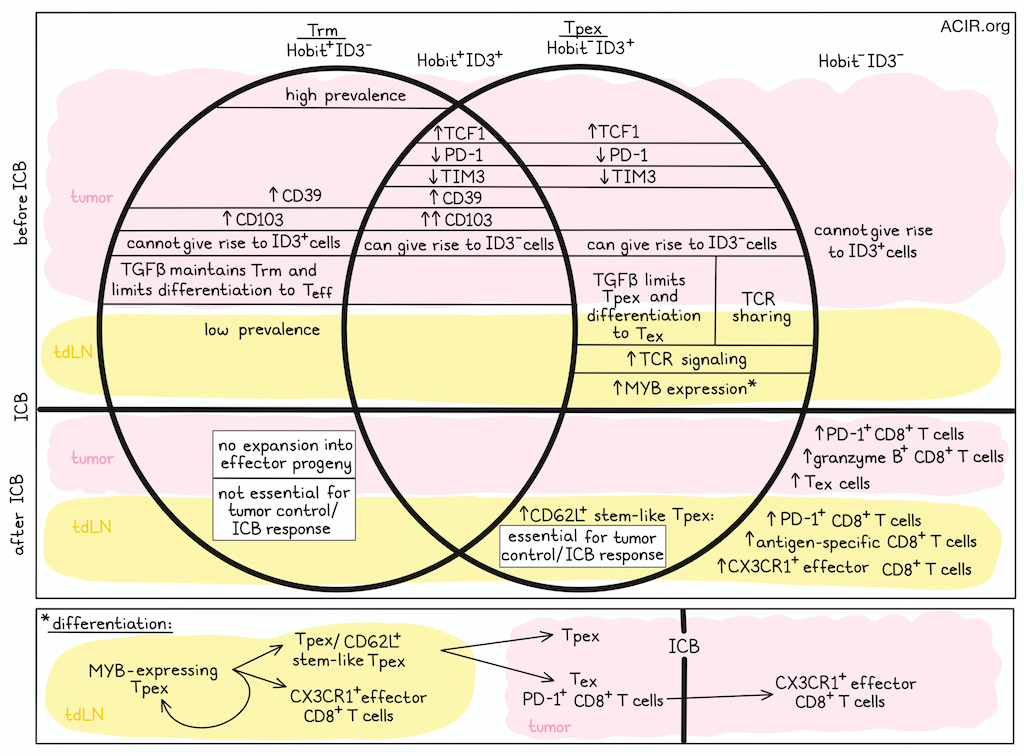
Reporter mice were used to track Tpex cells (ID3+) within the tumor microenvironment (TME). ID3+CD8+ cells expressed TCF1 and lacked TIM3. A large proportion of ID3- and ID3+ cells expressed canonical Trm cell markers, suggesting Tex and Tpex can both acquire Trm-like phenotypes. Another reporter model, using Hobit expression to mark Trm cells, showed Hobit+ Trm-like CD8+ T cells were abundantly present in the tumor, while they were rare in the spleen and TDLN. Intratumoral Hobit+ CD8+ T cells expressed Trm as well as Tpex and Tex markers.
To allow for simultaneous assessment of Trm-like and precursor cells, the researchers generated Hobit and Id3 double reporter mice. Among tumor-infiltrating CD8+ T cells, ID3+Hobit- and ID3+Hobit+ cells expressed high levels of TCF1 and low levels of PD-1 and TIM3. Both Trm subsets (Hobit+ID3+/-) expressed CD39, and Hobit+ID3+ cells expressed the highest levels of CD103. Tpex cell-specific genes were enriched in ID3+Hobit- subsets, and ID3+Hobit+ cells were enriched for both Tpex and Trm signatures. When four populations were sorted based on ID3 and Hobit expression and intratumorally transferred to recipient mice, both ID3+Hobit- and ID3+Hobit+ cells gave rise to ID3- populations, consistent with precursor characteristics, while Hobit+ID3- and Hobit-ID3-cells could not acquire ID3 expression.
Treating tumor-bearing ID3/Hobit double-reporter mice with ICB led to tumor regression and increased PD-1+ and granzyme B+ CD8+ T cells in the TME, which were mainly ID3-Hobit- cells. The researchers then performed fate mapping of Hobit-expressing cells and their progeny to determine whether Trm-like cells respond to ICB. After treatment, most expansion was among the non-labeled subset, suggesting effector progeny were mainly derived from non-Trm-like CD8+ T cells. To determine if Hobit+ Trm-like cells contributed to tumor control, mice were generated in which Tcf7 (TCF1) was deleted from Hobit+ cells. In this model, tumor control was not impacted, while TCF1 deletion from all CD8+ T cells resulted in reduced tumor control. In a model in which Trm-like cells were depleted from tumors, but not TDLNs, no difference in tumor response to ICB was observed. These data suggest that Hobit+ Trm-like CD8+ T cells are not essential for tumor control and ICB response.
The researchers then assessed TDLN Tpex cells, which were enriched for a TCR-signaling transcriptional signature, with extensive TCR sharing between these populations and the tumoral Tpex and Tex cells. Trajectory analyses showed that the TDLN cells gave rise to CX3CR1+ effector CD8+ T cells in the TDLN and Tex cells in the tumor, while maintaining the TDLN Tpex population.
In the TDLN, ICB induced expansion of PD-1+ and antigen-specific CD8+ T cells, including CD62L+ (an adhesion molecule critical for T cell migration) Tpex cells, as well as tumor-responsive CX3CR1+ effector cells, which were also detected in the periphery. Their importance for ICB response was confirmed by use of FTY720, which restricts T cell migration leading to retention in the TLDN, as this resulted in abrogation of ICB efficacy.
The researchers then studied the transcription factor MYB, which has been associated with differentiation and maintenance of CD62L+ stem-like Tpex cells in chronic infection. Tpex cells in the TDLN, but not in the tumor, expressed high levels of MYB. In mice containing MYB-deficient CD8+ T cells, tumor growth was accelerated, CD62L+ Tpex cells were lost in the TDLN, and there was a strong reduction of PD-1+CD8+ T cells and Tpex cells in the tumor. MYB-deficient PD-1+CD8+ T cells could not expand and generate CX3CR1+ effector CD8+ T cells in response to ICB. Together, these results pointed to the importance of MYB+ stem-like Tpex cells in the TDLN.
Since TGFβ is involved in both Tpex and Trm cell differentiation, its role on T cell fate was assessed. TGFβ-induced transcriptional signature enrichment was detected within the tumor and among a subset of TDLN Tpex cells. Mice in which TGFβ receptor-mediated signaling was abrogated in CD8+ T cells (by CD8+-specific knockout of Tgfbr2, the TGFβ receptor) were able to control tumors, with more CD8+ T cells infiltrating the tumor. These were mostly PD-1+CD8+ T cells, with few TCF1+ Tpex and a loss of CD103+ Trm-like cells. In the TDLN, PD-1+CD8+ and CD62L+ Tpex cells increased, while CD103+CD69+ Tpex cells decreased. CX3CR1+ Tex cells increased in both the blood and TDLN. Thus, TGFβ has opposing roles dependent on the target cell, limiting stem-like Tpex and differentiation to Tex cells in the TDLN and the tumor, while being required for maintenance of Trm-like cells in the tumor.
In mice in which Tgfbr2 was absent from Hobit+ cells, tumor control improved, with increases in PD-1+TIM3+ IFNγ/TNF/GZMB-producing CD8+ T cells in the tumor. Few T cells had tissue-residency features. Fate mapper analyses showed that removal of TGFβ signaling in developing Trm-like cells resulted in the formation of an effector phenotype with improved functionality, suggesting TGFβ drives maintenance of the Trm phenotype in tumors, and loss of TGFβ signaling shifts Trm cells to an effector phenotype.
To determine whether these data might also apply to human T cells, the researchers assessed TCGA data, which showed a correlation between TGFB1 transcripts and a Tpex signature. Further, in samples from patients with non-small cell lung cancer, the frequency of CD127+CD103+ Tpex cells correlated with tumoral TGFB1 mRNA expression. In tumors, CD127+CD103+ and CD127+CD103- subsets were detected within the CD39+CD8+ Tpex population, and in breast cancer TDLNs, CD127+CD103- stem-like Tpex cells were more abundant. CD8+ Tpex and Tex cells from lung cancer samples were stimulated with anti-CD3/CD28 with or without TGFβ. This revealed that TGFβ induced CD103, PD-1, and CCR7 on Tpex cells, while it downregulated GZMB and TIM3. These cells were transcriptionally similar to conventional Tpex cells, while cells stimulated in absence of TGFβ were more similar to stem-like Tpex cells. Thus, consistent with the results in the mouse, TGFβ is important to inducing the Trm phenotype while limiting stem-like Tpex and their differentiation to effectors.
Together, these data suggest that tumor Trm cells are not required for tumor control and ICB efficacy, and are restricted from becoming effector cells through tumoral TGFβ. MYB-dependent CD62L+ Tpex cells in the TDLN, on the other hand, are essential for antitumor responses and generate CX3CR1+ effector cells that migrate to tumors and induce responses to ICB. These data provide new clues as to which cells to target with immunotherapies to improve the outcomes of current treatment strategies.
Write-up by Maartje Wouters, image by Lauren Hitchings



