Last week, the ACIR team attended the virtual 35th Anniversary Annual Meeting of the Society for Immunotherapy of Cancer (SITC). This week’s extensive special feature covers selected talks from the conference. We have organized the content by topics below.
- Immune checkpoint blockade
- Jim Allison
- Ira Mellman
- Nils Rudqvist
- Kurt Jenkins
- Big data
- Matt Spitzer
- Yardena Samuels
- Anastasia Prokopi
- Arlene Sharpe
- Clinical trials of combinatorial therapies
- Adi Diab
- John Thompson
- Elizabeth A. Mittendorf
- Macrophages
- Jennifer L. Guerriero
- David Sallman
- Tumor microenvironment
- Elizabeth M. Jaffee
- Shannon J. Turley
- Pablo Umana
- Cancer vaccines
- Cathy Wu
- Kristen Radford
- NK cells and T cell therapies
- Helen E. Heslop
- Andreas Lundquist
Immune checkpoint blockade
Immune Checkpoint Blockade in Cancer Therapy: New insights into the therapeutic mechanisms of anti-CTLA-4 and anti-PD-1- James P. Allison, PhD - The University of Texas MD Anderson Cancer Center
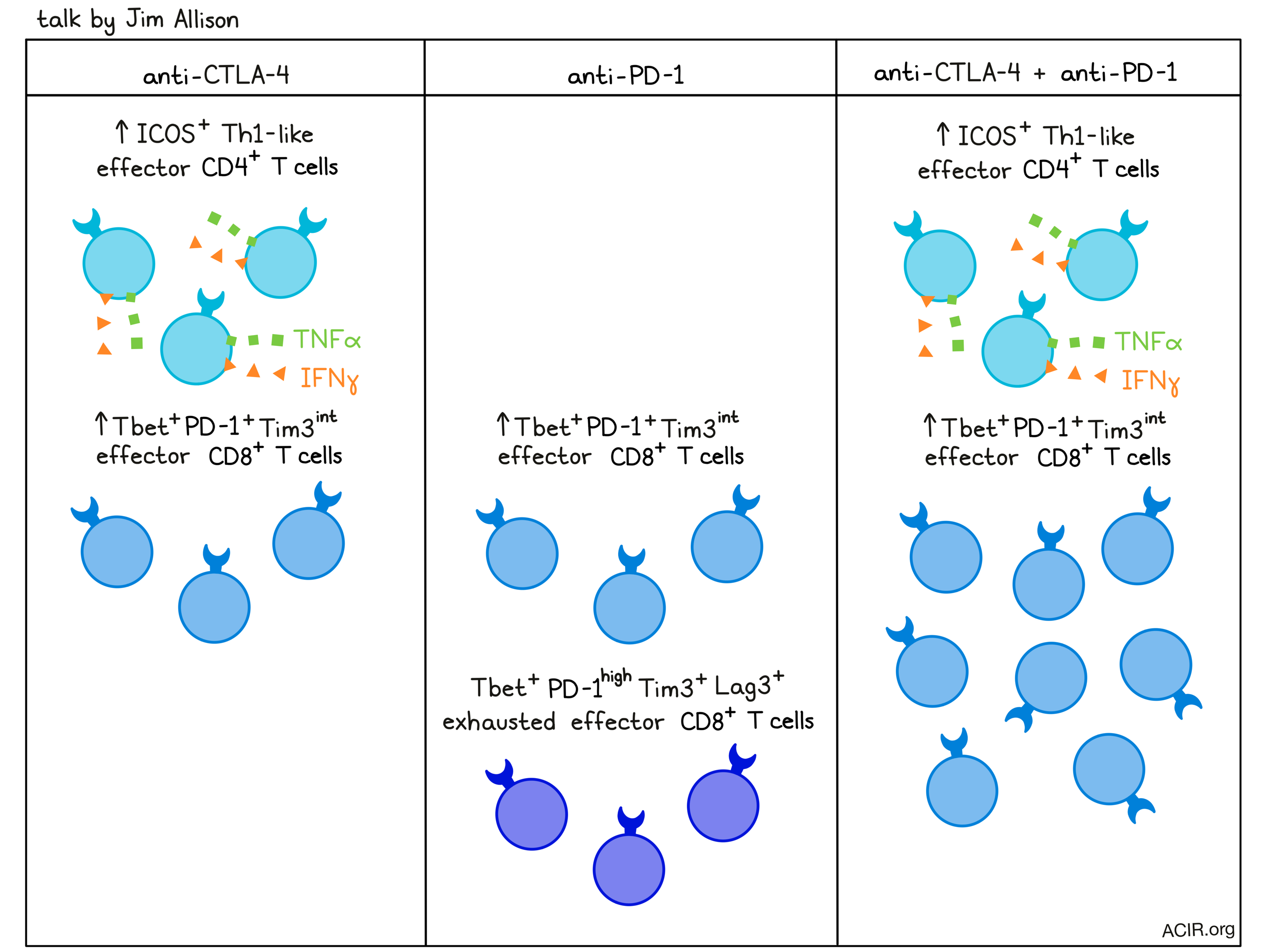
Pioneer of cancer immunotherapy and Nobel laureate Jim Allison gave a talk on T cell phenotypes that contribute to the efficacy of immune checkpoint blockade, and how the most common checkpoint blockades, anti-CTLA-4 and anti-PD-1, work alone and in combination. To begin, he discussed how tumors get a head start over the T cell response, as the tumor microenvironment provides TCR stimulation, but not CD28 costimulation. This allows tumors to go unnoticed until dendritic cells move in on dying tumor cells and provide T cells with the costimulatory signals necessary to initiate an antitumor response. Further, CD28 costimulation is hampered by the CTLA-4 on T cells, which competes with CD28 for engagement and sends inhibitory rather than activating signals. Blocking CTLA-4, which allows for continued proliferation, can enhance T cell priming, increase T cell infiltration into tumors, and expand clonal diversity. Recent research conducted by Dr. Allison and Spencer Wei showed that anti-CTLA-4 therapy expanded a population of ICOS+ Th1-like effector CD4+ T cells that produced IFNγ and TNFα and were essential to antitumor efficacy. This population was interesting, as ICOS expression is usually associated with follicular T helper cells or Tregs. Knockout of CTLA-4 also generated cells with this novel phenotype, as well as several other atypical CD4+ T cell phenotypes, suggesting that CTLA-4 constrains CD4+ T cell differentiation in concert with environmental cues and TCR signal strength. In addition to its effects on CD4+ T cells, anti-CTLA-4 therapy also induced a population of Tbet+EOMES+ terminally differentiated effector CD8+ T cells. Blocking PD-1, which is understood to act on more differentiated cells, expanded this same subset of Tbet+EOMES+ terminally differentiated effector CD8+ T cells, along with a population of Tbet+PD-1highLag3+Tim3+ exhausted effector CD8+ T cells. The latter population required the continued presence of anti-PD-1 for continued proliferation, which may explain the need for extended dosing of PD-1 axis-targeting agents. Investigating how anti-CTLA-4 and anti-PD-1 would affect T cell phenotypes in combination, Allison and colleagues found that combined checkpoint blockade greatly increased ICOS+Tbet+ Th1-like effector CD4+ T cells (like in CTLA-4 monotherapy) and Tbet+EOMES+KLRG1+ CD8+ effector T cells (like in either anti-CTLA-4 or anti-PD-1 monotherapy). Interestingly, while anti-PD-1 monotherapy increased populations of phenotypically exhausted T cells, the combination with CTLA-4 brought the same population down, but increased the non-exhausted PD-1+Tim3int population. In ongoing research, Allison has been investigating what happens to the exhausted cells, and put forward the hypothesis that exhaustion of effector cells may be prevented in the presence of the continued CD28 costimulation that occurs in the context of CTLA-4 blockade. Allison believes that better understanding the mechanisms behind combination therapy is critical to optimally treating patients.
Reassessing the mechanism of checkpoint inhibition - Ira Mellman, PhD - Genentech
After leading off his talk with a short video and imaging from a high resolution electron microscopy study of a T cell–tumor cell interaction highlighting the structural complexity of the immunological synapses, Ira Mellman discussed how many early understandings of the mechanisms underpinning the success of checkpoint blockade efficacy may not be entirely accurate, or may not represent the full scope of what actually happens and where. For example, while CTLA-4 blockade is typically thought to work during priming in the lymph nodes, there is evidence (in mice, but not in humans) to suggest that it may also impact Treg populations in the tumor bed. Similarly, while PD-1/PD-L1 blockade has previously been understood to work by reinvigorating exhausted T cells in the tumor bed, recent work has shown that it may play a more significant role earlier on, impacting T cell stimulation and expansion in secondary lymphoid organs. In recent studies, Mellman and his colleagues, along with others, have found that PD-1 may regulate the CD28 costimulatory pathway rather than the TCR pathway. Further, PD-L1 was previously understood to be expressed by tumor cells as a defense mechanism; however, recent studies have shown that PD-L1 on immune cells in the tumor microenvironment plays a more important role in dictating anti-PD-1 or anti-PD-L1 efficacy, and in some cases, is predictive of response. Looking more closely at specific immune cells expressing PD-L1, researchers have found that while macrophages in the tumor microenvironment harbor ten times more PD-L1 compared to dendritic cells and are the major source of PD-L1 in the TME, PD-L1 on a per-cell basis was more abundant on dendritic cells and contributed much more to hindering antitumor effects. Further, PD-1/PD-L1 interactions in this setting did not appear to drive T cell exhaustion. Instead, blocking the PD-L1 axis induced expansion of stem-like memory T cells, which are antigen committed and can give rise to memory, effector, and exhausted T cells. Along with PD-1, these stem-like memory T cells co-express TIGIT, a negative regulatory molecule that competes with costimulatory CD226 for interactions with their ligand, CD155. While it was previously observed that TIGIT and CD226 competed for CD155 in a manner analogous to CTLA-4 competing with CD28 for binding to CD80/86, recent studies have shown that PD-1 directly impacts the phosphorylation state of CD226, indicating that CD226 is coordinately regulated by both TIGIT and PD-1. Early clinical studies suggest there may be a benefit to blocking both pathways. Evidence is stacking up to show that blocking PD-1/PD-L1 does little to reverse T cell exhaustion:
- epigenetic signatures of exhaustion are essentially locked in;
- anti-PD-1 therapy is just as effective when selectively delivered in lymph nodes and excluded from the tumor bed;
- new TCR clonotypes appear following therapy, and new clonotypes in both blood and the tumor correlate with response, more so than on pre-existing T cell responses; and
- antitumor responses depend on T cells newly trafficked from lymphoid organs.
As more patients are deriving clinical benefits from checkpoint blockade, the research community is learning more about the basic immunology that underpins these successes. With more information on hand, paradigms are shifting, and our understanding of checkpoint blockades must follow where the evidence takes us, even if it challenges current understandings and assumptions.
Radiotherapy and CTLA-4 blockade expand anti-tumor T cells differentiation states and cooperate with CD40 agonist to induce tumor rejection - Nils Rudqvist, PhD - The University of Texas MD Anderson Cancer Center
Leveraging some encouraging clinical results of hypofractionated radiotherapy (RT) combined with anti-CTLA-4 (ipilimumab) in NSCLC, Nils Rudqvist ‘reverse-translated’ these findings to uncover a new approach to improve the combination. Seeing that RT + ipilimumab altered the T cell repertoire, including inducing clonal expansion of T cells with new antigen specificities, Rudqvist turned to the murine 4T1 TNBC model to mimic the combination therapy, observing comparable increases in T cell clonality and tumor infiltration. Drilling deeper by analyzing single-cell RNA sequencing of T cells in infiltrated tumors, and backing it up with bulk gene signature analysis, he observed the frequencies and absolute abundances of 3 populations changed significantly in response to the combination therapy. Analysis of the key differentially expressed genes revealed an increase in a CD4+ population expressing CD40 ligand and an increase in two CD8+ populations (with a relative shift from a cytotoxic type expressing GzmB and Perforin to a polyfunctional T cell expressing IFNγ and TNFα). As the cytotoxic CD8+ T cells expressed PD-1 and LAG3, and the CD4+ T cell population expressed CD40 ligand, targeting these pathways with antagonistic antibodies to PD-1 and LAG-3, and an agonistic antibody (Ab) to CD40 (to enhance the activity of CD40-expressing dendritic cells), seemed promising. Indeed, agonist CD40 Ab combined with RT and anti-CTLA-4 dramatically improved activity in the 4T1 model, leading to complete responses, which had never been observed with RT plus anti-CTLA-4. Neither anti-PD-1 nor anti-LAG3 improved antitumor efficacy in this model. These findings open a clear clinical opportunity for a triple combination study (RT + ipilimumab + agonistic CD40 Ab) and highlight the importance of dendritic cell activity and T cell phenotypic markers beyond PD-1.
Tumor-activated Fc-engineered anti-CTLA-4 monoclonal antibody, XTX101, demonstrates tumor-selective PD and efficacy in preclinical models - Kurt Jenkins, PhD - Xilio Therapeutics
Aiming to improve both the efficacy and safety of ipilimumab, the first approved modern immunotherapy, Kurt Jenkins described XTX101, an anti-CTLA-4 antibody with enhanced affinity for CTLA-4, increased ADCC activity via Fc-engineering, and tumor selectivity via a protease-releasable CDR binding peptide. Tumor selectivity, a key to capitalizing on the enhanced binding and functionality of the antibody, was achieved by first identifying a peptide that bound to the CDRs of the parent antibody and blocked CTLA-4 binding, and then using structural modeling to find an appropriate peptide linker to covalently link the peptide to the N-terminus of the light chain. In theory, the inert antibody could then be selectively activated by the enhanced protease activity in the tumor microenvironment environment. In vitro studies demonstrated a matrix metalloprotease-dependent switch in binding capability, and improved function in competitive ELISA (with CD80 and CD86) and cellular assays (IL-2 production and ADCC activity). Importantly, in vivo, XTX101 showed tumor clearance in the MD49 model at 1/10 of the dose of a murine ipilimumab analog. Mechanistically, tumors in animals treated with XTX101 showed higher CD8+ T cell infiltration, greater Treg depletion. Less potent stimulation of CD4+ T cell proliferation was observed than in animals treated with the ipilimumab analog. To examine whether diverse cancer types possess a suitable proteolytic environment, 85 samples from 8 different human cancer types were briefly cultured, and the culture supernatants were assessed for their ability to activate XTX101 by peptide release. 65% of the 85 samples activated XTX101, suggesting broad suitability across and within cancer types and supporting clinical testing.
Big data
Understanding immune responses to cancer using single cell data - Matt Spitzer, PhD - University of California, San Francisco
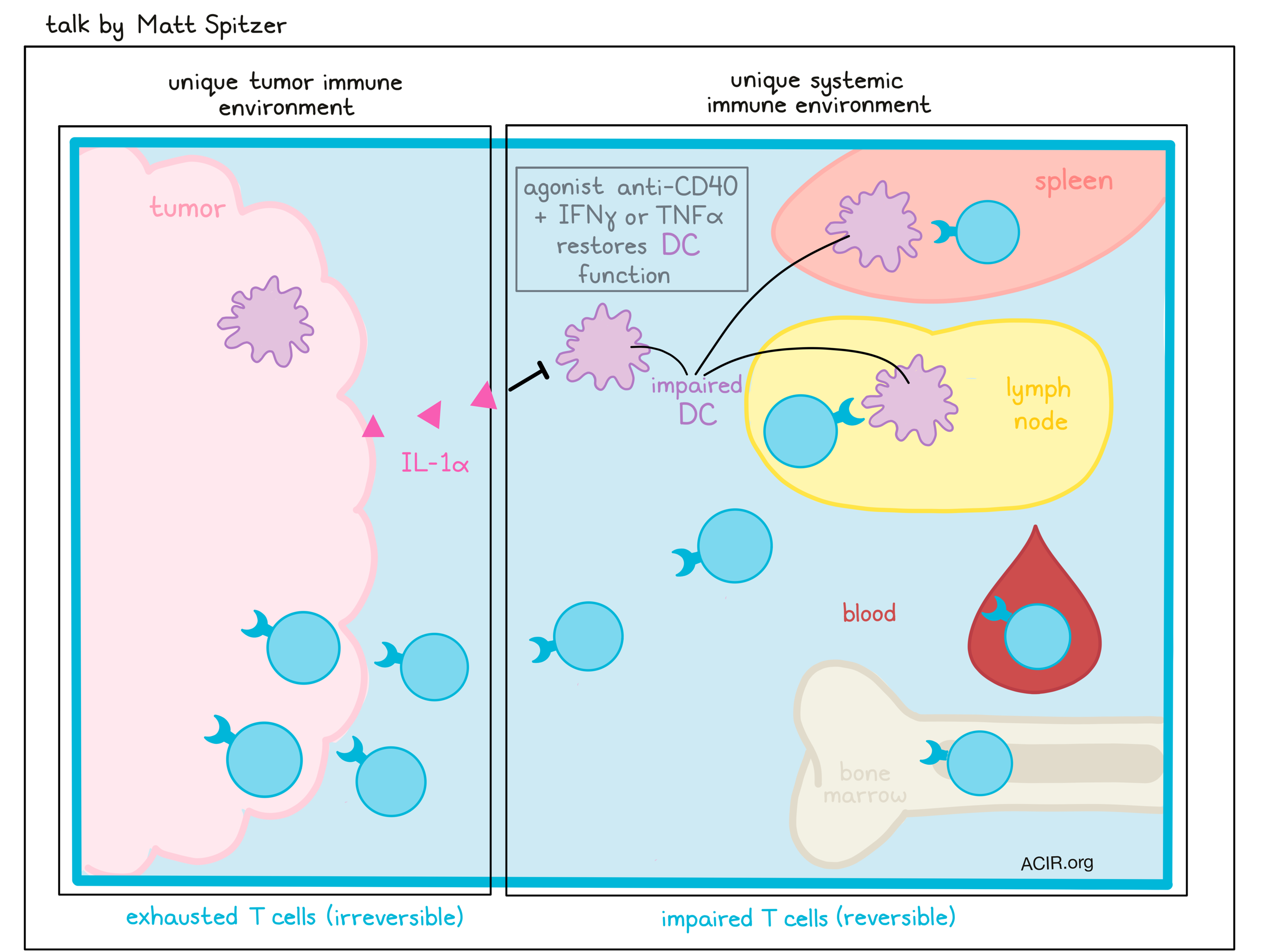
Zooming out from what happens in the tumor microenvironment, Matt Spitzer’s talk focused on what happens systemically when cancer develops. To study this, Spitzer has used CYTOF to analyze immune cells at the single-cell level from tumor tissue and various immune populations in lymph nodes, spleen, bone marrow and blood. In a tumor model resistant to anti-PD-1 therapy, Spitzer and colleagues had previously found that antibodies surrounding transplanted tumors could aid in the activation of DCs, and that the addition of CD40 agonist with IFNγ or TNFα could further enhance activation and induce antitumor responses in T cells, which were already enhanced with anti-PD-1. In a setting of effective therapy, immune cell proliferation was sustained outside of the tumor, particularly in cases in which therapy was most effective. Further investigation showed that a systemic immune response and cells coming from outside of the tumor were required for tumor eradication, including in anti-PD-1 therapy. Curious as to whether the tumor impacts systemic immune responses, Spitzer and colleagues profiled different mouse tumor models and found that each developed distinct intratumoral and systemic immune cell compositions compared to other models or control mice. Looking specifically at T cells, they identified systemic, tissue-specific changes in T cell phenotypes with progressing tumor burden. A particular population of exhausted T cells accumulated uniquely in the tumor and were irreversibly impaired by epigenetic changes. Asking whether changes induced by the tumor would apply to newly induced T cells, Spitzer and colleagues infected tumor-bearing or control mice with LCMV or Listeria. T cell responses were impaired in magnitude and functionality, but co-transfer experiments indicated that T cell impairment outside of the tumor was less permanent and could be reversed in a different physiological context. DCs were also found to be systemically impaired, suggesting that T cell impairment in tumor-bearing mice might be the result of impaired T cell priming. While T cell-targeted therapies like IL-12 failed to rescue deficient T cell responses, the addition of agonist anti-CD40 for DC stimulation restored systemic T cell responses. Looking at tumor secretions that might impair DC function, Spitzer and colleagues found that tumor expression of IL-1α affected systemic DC function. Another group found similar results when their tumor model expressed IL-6, suggesting that different tumors may utilize different defense mechanisms to dampen antitumor immune responses. Surgical resection of the tumors in these cases was sufficient to restore de novo systemic immune responses, highlighting the flexibility of the immune system and its ability to recover.
Capturing heterogeneity and the HLA-presented landscape in melanoma - Yardena Samuels, PhD – Weizmann Institute of Science
Acknowledging the difference in the predictive value of tumor heterogeneity and mutational load as biomarkers of checkpoint blockade response, Yardena Samuels’ group created a model system to study these factors in-depth. By exposing mouse melanoma cells to UVB, the researchers created aggressive, polyclonal tumor cell populations with high heterogeneity and increased mutational load. From these tumors, single cell lines were derived to create homogenous populations with variability in mutational load; these lines were rejected quickly in immunocompetent mice. These homogeneous tumors were more highly infiltrated with IFNγ+CD137+granzyme B+ CD8+ T cells, while heterogeneous tumors had fewer CD8+ T cells, which were restricted to the margins. Using HLA peptidomics, 17 neoantigens were identified, some of which mediated tumor cell killing. A phylogenetic tree could be constructed from the mutational data and by combining different cell lines from distinct parts of the tree to induce tumors, the researchers found that the number of clones and their genetic diversity both played important roles in mediating tumor growth and rejection. They also showed in human melanoma that mutational diversity correlated with response to checkpoint blockade. Looking further into what peptides are presented by tumor cells, bacteria-derived, HLA-presented peptides were identified from bacterial species found inside human melanoma cells, some of which were recurrent and immune-reactive. Lastly, Samuels presented new work on the identification of IFNγ-induced ribosomal frame-shifting and the subsequent HLA presentation of aberrant peptides. Reactive T cells in the tumor produce IFNγ, inducing the production of IDO1 in tumor cells, which results in tryptophan degradation, but the consequences on protein translation are not known. Using riboseq and mRNAseq on melanoma cell lines exposed to IFNγ, the researchers were able to show that the ribosome stalled at internal tryptophan codons, resulting in frameshifts, and in some genes 15 codons later, possibly due to a structurally disordered peptide translated from the out-of-frame sequence and resulting in quality control-induced degradation. Some of these out-of-frame peptides were then shown to be presented by HLA on the cell surface, and some were immunoreactive. These data suggest a new layer of opportunity for tumor-targeting T cells and also expanded tumor heterogeneity. An important remaining question is whether these novel peptides help or distract the antitumor response.
High dimensional analysis of the human lymph node during melanoma progression reveals shifts in myeloid content that relate to differential T cell content - Anastasia Prokopi, PhD - Amsterdam UMC
Acknowledging the importance of the sentinel lymph node (SLN) in melanoma as the site where the antitumor immune response initiates, but also the first site of metastasis, Anastasia Prokopi presented immunophenotyping data of the SLN at various stages of disease. Using mass cytometry and multi-parameter flow cytometry, myeloid and T cells were characterized in healthy, tumor-negative SLNs, tumor-positive SLNs, and SLNs with extensive metastatic lesions (LNmet). Melanoma progression induced major immunological changes, including an overall decrease in CD45+ immune cells, with specific increases in B cells, NK cells, granulocytes, and multiple myeloid cell populations, and a specific decrease in T cells. More detailed analysis of the myeloid cells revealed a decrease in DCs, both CD141+ cDC1 and cDC2, and an increase in a CD11c+HLA-DR+CD14+ subsets expressing markers such as CD163 and PD-L1/L2, suggestive of an immunosuppressive phenotype. Using healthy donor monocytes that were matured and treated with melanoma supernatants, the researchers confirmed this CD14+ phenotype was monocyte-derived. During progression, there was a gradual decrease in naïve T cells, and Treg and Trm cells acquired checkpoint molecules (TIGIT and PD-1). The changes in the T cell compartment correlated with changes in the myeloid cells, suggesting myeloid cells might serve as a target for therapy to ensure adequate T cell differentiation and improve immune checkpoint therapy efficacy.
Discovery of new immunotherapy targets and mechanisms leveraging CRISPR - Arlene H. Sharpe, MD, PhD - Harvard Medical School
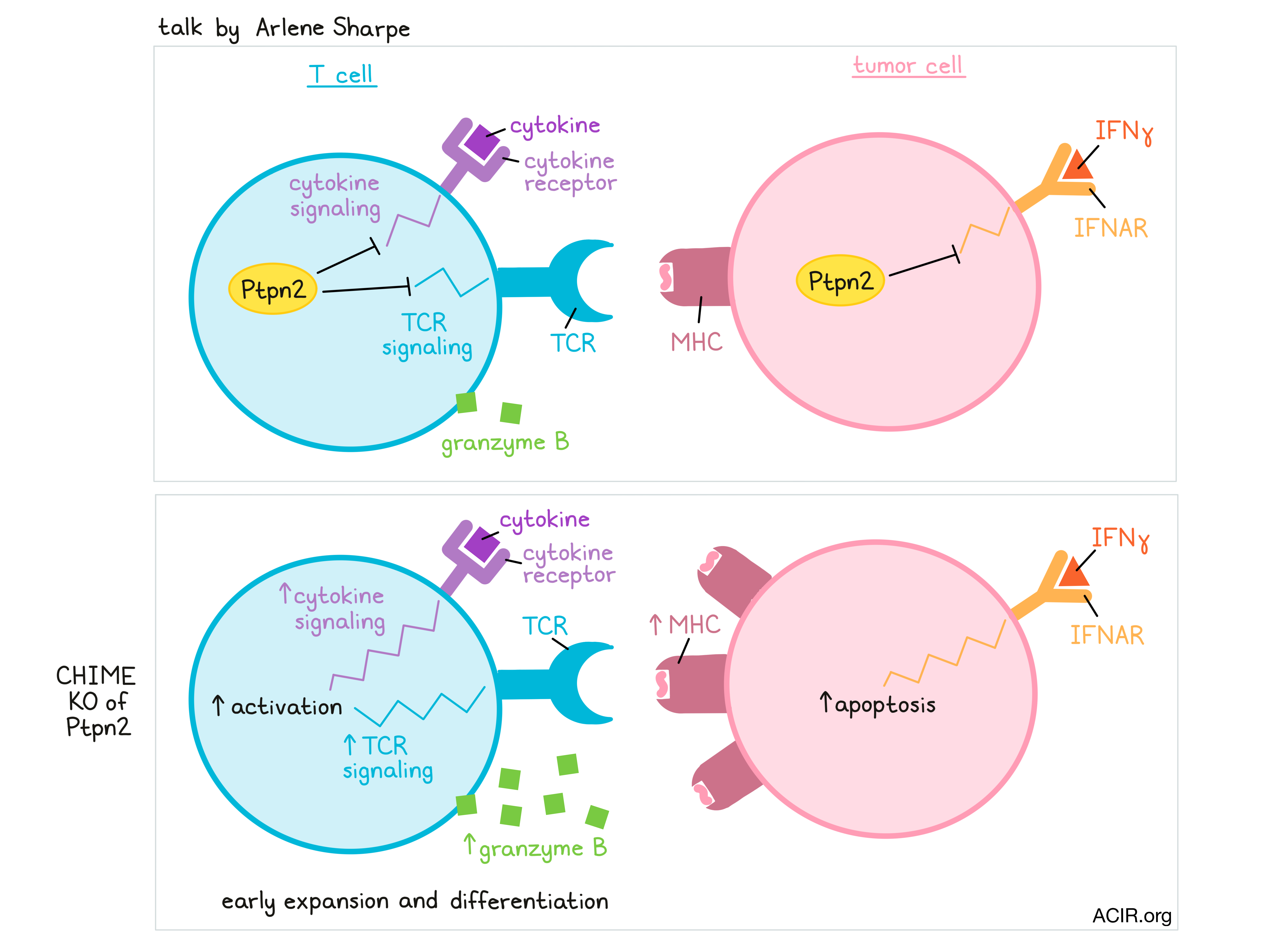
In her talk, Arlene Sharpe discussed the development of CHIME – a CRISPR-based platform that allows researchers to knock out genes in immune lineages (B cells, naive lymphocytes, CD4+ T cells, CD8+ T cells, macrophages, and DCs) in vivo, without affecting immune homeostasis or function. CHIME can even be used to evaluate genes in a pooled fashion and is being developed to knock out two genes simultaneously in the same cell. This system allows for the identification of immune cell-intrinsic effects and known and novel regulators of antitumor responses. In one screen, a novel regulator, Ptpn2, caught Sharpe and her colleagues’ attention. Ptpn2 is a phosphatase that plays a role as a negative regulator of TCR signaling and cytokine signaling in T cells, and is associated with autoimmunity in both mice and humans. Further, it also plays a cell-intrinsic role in tumor cells, regulating IFNγ signaling and MHC expression, thus protecting tumor cells. Evaluating Ptpn2 using the CHIME system, Sharpe found that deletion of Ptpn2 in T cells promoted early expansion, induced increased granzyme B production, and provided a competitive advantage over wild-type T cells during LCMV infection. Investigating whether Ptpn2 played a role in controlling subpopulations of exhausted cells, Sharpe found that Ptpn2 deletion led to an increase in terminally exhausted T cells (highly cytotoxic, but less persistent and less proliferative) by releasing the brakes on type I IFN signaling, thus enhancing early differentiation of this population. Ptpn2 deletion did not affect the numbers of progenitor exhausted T cells. In a tumor setting, Ptpn2 deletion in CD8+ T cells improved cytotoxic responses (as it did in the LCMV model), and Ptpn2 deletion in the entire hematopoietic compartment induced total clearance of MC38 tumors, dependent on the presence of CD8+ T cells. Using B16 tumors to evaluate whether this strategy might synergize with PD-1 blockade, Sharpe and colleagues showed that while blockade of PD-1 showed no effect as a monotherapy in this model, it enhanced T cell cytotoxicity and induced notable antitumor effects in mice with Ptpn2 knocked out in hematopoietic cells.
Clinical trials of combinatorial therapies
REVEAL: Phase 1 Dose Escalation Study of NKTR 262, a Novel TLR7/8 Agonist, Plus Bempegaldesleukin: Local Innate Immune Activation and Systemic Adaptive Immune Expansion for Treating Solid Tumors - Adi Diab, MD - University of Texas MD Anderson Cancer Center
Aiming to activate both innate and adaptive immunity, Adi Diab discussed preliminary results of a phase 1 dose-escalation trial combining the intratumoral delivery of a TLR7/8 agonist (NKTR-262) with intravenous Bempegaldesleukin (BEMPEG), a CD122-preferential IL-2 pathway agonist. The combination was tolerable, but four patients discontinued treatment due to adverse events. No Recommended Phase 2 Dose (RP2D) was reached, so the highest dose was defined as the RP2D. Of the 28 patients evaluable for efficacy, eight (29%) showed regression of the injected lesion. In heavily pretreated melanoma, 9% (2/22) experienced an objective response. Patients with melanoma had increased CXCL10 (IP-10) and dose-dependent induction of type I IFN genes, which correlated with the density of CD11c cells in biopsies, consistent with TLR 7/8 engagement. Additionally, these patients had increased peripheral proliferation of T (both CD4+ and CD8+) and NK cells at cycle 2, the first cycle of combined NKTR-262 and BEMPEG. These findings support ongoing phase IB and II trials, including an arm that adds Nivolumab to the combination.
Phase I/Ib first in human study of NIZ985 (HetIL 15; IL 15/IL 15Rα) alone and in combination with spartalizumab, in adults with advanced and metastatic solid tumors - John Thompson, MD - University of Washington Seattle Cancer Care
Intending to induce T and NK cell responses while blocking the PD-1 pathway, John Thompson presented data from a phase I dose-escalation/expansion trial evaluating the safety and efficacy of subcutaneous NIZ985, a recombinant heterodimer of IL-15 and IL-15Rα (hetIL-15) and intravenous spartalizumab, a humanized IgG4 anti-PD-1 antibody. Heavily pretreated patients with metastatic or unresectable solid tumors were included in this trial with single-agent NIZ985 or the combination strategy. No dose-limiting toxicities were observed, with 6/36 patients having treatment-related serious adverse events. Pharmacokinetic studies showed that serum concentration was dose-dependent, and repeat dosing suggested that NIZ985 created an IL-15 sink. NIZ985 induced proliferation of peripheral CD8+ T and NK cells, but no correlation was found between this expansion and clinical response. The researchers also observed an increase in plasma levels of the inflammatory cytokines IFNγ, IL-18, IL-15, and IL-12 P40. While the single treatment with NIZ985 had limited efficacy (30% stable disease; no partial responses), the combination treatment resulted in partial responses in three patients (5%) and SD in 15 patients (27%).
Neoadjuvant Breast Cancer - Elizabeth A. Mittendorf, MD, PhD – Dana-Farber Cancer Institute / Brigham & Women's Cancer Center
Invigorated by the success of checkpoint therapy with atezolizumab in the IMpassion130 trial in triple negative breast cancer (TNBC), a series of studies in the neoadjuvant setting for breast cancer have been initiated. Some of the rationale and the results of these were reviewed by Elizabeth Mittendorf. Immunohistochemistry analysis of breast cancer samples typically show a higher tumor-infiltrating cell count and higher PD-L1 levels in primary tumors than at metastatic sites – markers of increased benefit with anti-PD-1 axis therapy. Five exploratory neoadjuvant trials were reported on, and although the trials differ in the chemotherapy backbone used, the target (PD-1 or PD-L1) and the stage/subtype of disease, a few observations are noteworthy. In several trials, a clear benefit in pathological complete response rate (pCR) was observed, which typically was independent of PD-L1 status and sometimes varied by disease stage. Chemotherapy backbones containing anthracyclines may be most effective, and although event-free survival periods were extended, whether the addition of immunotherapy translates to longer-term survival is still a big unknown and awaits longer follow-up. Adrenal insufficiency emerged as an unanticipated adverse event (6.7%) in the paclitaxel + pembrolizumab arm compared to 0% in the chemotherapy alone arm of the I-SPY2 trial, with rates just below 3% in two other trials utilizing neoadjuvant checkpoint blockade. This potentially irreversible impact would complicate use in a population, some of whom might be cured. Overall, the results are encouraging, but many questions remain (sequencing of therapies, follow-up with adjuvant immunotherapy, de-escalation of chemotherapy doses, among many others), including, importantly, whether it is needed in early stage TNBC, where patients with pCR following neoadjuvant chemotherapy have a very good prognosis.
Macrophages
Differentiating Macrophages are Regulated by PARP Inhibitors and can be Harnessed to Overcome PARP Inhibitor Resistance in BRCA Associated Triple Negative Breast Cancer - Jennifer Guerriero, PhD - Dana-Farber Cancer Institute
Jennifer Guerriero discussed the search for rational therapeutic targets for tumor-associated macrophages (TAMs). In BRCA-associated TNBC, excess cytosolic DNA activates the cGAS/STING pathway, leading to chemokine production and immune cell recruitment. PARP inhibitors, inducing further DNA damage in TNBC cells, lead to greater T cell recruitment and antitumor efficacy. However, clinical response to PARP inhibitors and checkpoint blockade has not demonstrated superior activity over PARP inhibition alone, despite high T cell infiltration. Guerriero’s group assessed whether TAMs might limit this therapeutic response using a murine BRCA1- and TP53-deficient GEMM model. Treatment after tumors emerged with the PARP inhibitor olaparib resulted in increased T cell and TAM infiltration, which upregulated CD80, PD-L1, and CSF1R, a phenotype that could be replicated in vitro with human monocytes matured in the presence of olaparib. CSF-1R regulates the proliferation, differentiation, and survival of mature macrophages. Treating the BRCA1-mutant mouse models with olaparib and anti-CSF1R increased survival. Immunophenotyping revealed increased numbers of TAMs, but with a less suppressive phenotype. There was an increase in granzyme B+ T cells and a decrease in Tregs. Depletion of T cells showed that both innate and adaptive immune responses were required for durable outcomes. To assess how the TAMs affected T cells, T cells were exposed to olaparib-treated macrophage-conditioned media, resulting in T cell function inhibition and apoptosis. Additionally, RNAseq implicated an upregulation of metabolic pathways in olaparib-treated macrophages, including the SREBP1-mediated fatty acid synthesis pathway. The addition of SREBP1 inhibition to the combination strategy increased efficacy even more.
CD47 Macrophage Checkpoint-Based Immunotherapy (Lymphomas, MDS, AML) - David Sallman, MD – Moffitt Cancer Center
Looking into checkpoint blockade that aids macrophage responses, David Sallman presented data on Magrolimab, a first-in-class macrophage checkpoint inhibitor targeting CD47. CD47 is the ligand to SIRPα and provides a ‘do not eat me’ signal to macrophages. CD47 is overexpressed by multiple tumor types. In acute myeloid leukemia (AML), CD47 is overexpressed on leukemic stem cells and bulk AML, resulting in a strong survival advantage of these cells, with expression predicting worse clinical outcomes. A phase II trial combining magrolimab and rituximab (anti-CD20) in non-Hodgkin lymphoma (NHL) resulted in an ORR of 45%, with a median time to response of 1.8 months. Magrolimab also synergizes with the chemotherapeutic agent azacytidine, as this agent induces ‘eat me’ signals, such as calreticulin, on cancer cells, inducing macrophage antitumor responses. In a phase Ib trial including 68 patients with high-risk MDS or AML, the combination of magrolimab and azacytidine was well tolerated with no significant adverse immune events. On-target anemia was induced, but improved over time on treatment. No deaths occurred in the first 60 days of study, with an ORR for MDS of 91%, with a 42% CR, and an ORR for AML of 64%, with a 56% CR. In 16 patients with mutant TP53, the ORR was 75%. The time to response was faster (1.9 months) and favorable over azacytidine alone, which historically has a CR rate of 6-17%. The median OS was not reached, suggesting deep and durable responses. Multiple other agents, such as SIRPα fusion proteins to decrease or eliminate red blood cell clearance, and combination modalities are currently being tested for myeloid neoplasms.
Tumor microenvironment
Turning Immunologically Quiescent Tumors into Immune Responsive Cancers - Elizabeth M. Jaffee, MD – Sidney Kimmel Cancer Center, Johns Hopkins University
In her Keynote presentation, Elizabeth Jaffee discussed how although new technologies have allowed for rapid development and an era of accelerated approvals in the field of cancer immunotherapy, there are still hurdles to overcome, including addressing immunosuppression in the TME, tumor heterogeneity, and ensuring a quality T cell response. Although multiple challenges exist, Dr. Jaffee believes that combination therapies will serve to maximize clinical benefits. Investigating pancreatic cancer, which is typically an immune ‘cold’ tumor type, Jaffee and colleagues have shown that vaccination with GVAX induces increased T cell induction and infiltration into tumors and subsequent upregulation of tumor defense mechanisms, like PD-L1 expression on tumor cells and, more importantly, on antigen-presenting cells. T cells in the tumor environment also undergo changes, increasing expression of inhibitory receptors like PD-1, and activating molecules like CD137. Past studies have shown that using anti-PD-1 further alters the TME, possibly by preventing exhaustion in T cells induced by the vaccine. More recently, Jaffee and colleagues wondered whether targeting T cell activity using agonists might also enhance antitumor responses. To this end, Dr. Jaffee and colleagues are conducting ongoing trials testing neoadjuvant and adjuvant CY/GVAX, anti-PD-1 antibody, and agonist anti-CD137 antibody in patients with resectable pancreatic cancer. While the addition of anti-PD-1 to the vaccine increased T cell infiltration into tumors, only with the addition of agonist anti-CD137 did the researchers observe evidence of partial tumor regression in some patients. Classification of functional states within CD8+ T cells showed that the triple combination therapy induced a greater number of amplified CD8+ T cell TCR clonotypes, most of which had a more cytotoxic phenotype (granzyme B+PD-1+). In clinical trials of GVAX plus ipilimumab, patients with pancreatic cancer showed responses to immunotherapy, although more delayed than desirable, given the aggressiveness of the disease. These responses could not be predicted by biomarkers at baseline, nor by early T cell activation (which occurred similarly across responders and non-responders following immunotherapy), suggesting that antitumor responses are hindered after the T cell activation phase. Together these trials suggested that the diversity and the magnitude of T cell response and the phenotype of those T cells were all important. A current trial investigating the use of Cy/GVAX priming followed by boosting with CRS-207 (a Listeria-based vaccine targeting mesothelin) or CRS-207 alone as vaccine strategies, both with ipilimumab plus nivolumab co-therapy, has shown promising reductions in the time to response, and increases in the rates and numbers of early responders. Further, researchers are for the first time seeing responses in the liver and effects in peritoneal masses. With continued investigation and exploration of rational combination therapies that target various challenges to antitumor immunity, perhaps even notoriously difficult-to-treat pancreatic cancers can be managed with immunotherapy.
Stromal cells in health, disease and immunotherapy - Shannon J. Turley, PhD - Genentech
Moving to the tumor stroma, Shannon Turley discussed work on cancer-associated fibroblasts (CAF). Acknowledging the role of fibroblasts in many diseases, her lab set out to define specific CAF subsets and therapeutic targets. Using scRNAseq comparing healthy and cancer tissues in mice, two major CAF subtypes were found: an IL-1-associated type (often represented as inflammatory; iCAFs), and a subtype associated with TGF-β signaling (TGF-β CAFs), comparable to myofibroblastic CAFs (myCAFs). TGF-β CAFs were enriched in extracellular matrix (ECM) remodeling, and the TGF-β CAF signature was associated with reduced survival and lack of response to checkpoint blockade, particularly in immune-excluded tumors. Targeting of this population by TGF-β suppression in combination with PD-L1 blockade in preclinical models of cancers with an immune-excluded phenotype induced changes in multiple cellular compartments. Remodeling took place in CAFs, resulting in reduced ECM production, relaxing of the ECM, and increases in antigen presentation and processing machinery. Memory precursor CD8+ T cells migrated into the tumor and expressed markers indicative of an activated state and ‘repressed’ CD8+ T cells were reduced. These effects were associated with improved response to therapy, as compared to PD-L1 or TGF-β blockade alone. Similar CAF populations were identified in humans, where a TGF-β CAF signature was associated with reduced survival in pancreatic cancer and limited response to anti-PD-L1 therapy in bladder cancer. Therefore, targeting of the stromal CAF compartment may help alleviate barriers for T cell infiltration to treat tumors with an immune ‘cold’ phenotype. Turley’s group went on to prepare an atlas of fibroblast cell types based on single-cell sequencing data across multiple normal and non-cancer diseased tissues, and observed pan-tissue, tissue-specific, and disease-specific subtypes, including observing the TGF-β CAF present in other diseased tissue.
PD1-IL2v: a next generation, PD-1-targeted IL-2R agonist for cancer immunotherapy - Pablo Umaña, PhD - Roche
Given that systemic IL-2 treatment has powerful therapeutic effects, but is limited in use because of major adverse events, there is a demand for engineering strategies to improve its therapeutic index. Pablo Umaña presented constructs that have an IL-2 variant (IL2v) that binds to and activates the dimeric IL-2Rβγ, abolishing CD25 (IL-2Rα) binding, improving tolerability, and avoiding preferential Treg activation. Combining the IL2v with tumor-targeting antibodies improves retention in the tumor. The first developed construct binds to stromal FAP, improves tolerability, and results in a robust expansion of peripheral CD8+ T and NK cells without affecting Tregs. Objective responses were achieved in patients who had not responded to previous checkpoint inhibition therapy. FAP-IL-2v is currently in phase IB/II trials in combination with checkpoint blockade. Since activated T cells also express CD25, their response to IL-2v is similarly impaired. To improve expansion of antigen-activated T cells, a fusion of IL-2v to a high-affinity anti-PD-1 antibody was prepared to bring the IL-2v to the surface of PD-1-expressing cells and enhance stimulation. The novel PD-1-targeted IL-2v fusion immunocytokine dramatically increased IL-2R signaling and was shown to bind in cis to PD-1+ TIL, delivering IL-2v to TIL that were potentially enriched for tumor specificity. In an aggressive orthotopic pancreatic cancer mouse model, this construct had a tumor eradication and survival benefit over FAP-IL2v combined with anti-PD-1. It led to a significant specific tumoral expansion of PD-1highgranzyme B+ highly active CD8+ effectors, while not affecting peripheral T cells. The human variant was found to be superior over anti-PD-1 and IL-2 combination treatment, and its efficacy could be enhanced by combining it with anti-PD-L1 or anti-cancer vaccines. The construct is currently entering clinical trials.
Cancer vaccines
Personal Cancer Vaccines: on the Path to Effective Cancer Immunity - Catherine J. Wu, MD - Dana Farber Cancer Institute
With a focus on neoantigens, Catherine Wu discussed how therapeutic cancer vaccines can be seen as an important adjunct to other therapies. Neoantigen-targeted vaccines can serve to steer an immune response against tumor cells by generating and expanding a tumor-specific T cell response with reduced toxicity towards normal, healthy tissue. In recent years, advances in next-gen sequencing approaches and HLA-binding prediction algorithms have allowed for the development of personalized vaccines specific for neoantigens that are only present in an individual patient’s cancer. Clinical studies in the adjuvant setting of high-risk melanoma and newly diagnosed glioblastoma have shown therapy with neoantigen-targeted vaccines (NeoVax) to be feasible, safe, and able to generate specific T cell responses; they also show signs of synergy with checkpoint blockade. Looking back at cryopreserved samples from patients who achieved durable responses to NeoVax with or without anti-PD-1 that persist to this day, Wu and her team identified transcriptional clusters and noted patterns in which T cells advance from naive, to cytotoxic, to memory states with little evidence of exhaustion. Further, as this pattern played out, dominant clonotypes persisted and T cell repertoires broadened, suggestive of epitope spreading. Current samples from the same patients show neoantigen-specific T cells persist long-term. In the case of a patient who had a recurrence shortly after vaccination, and a subsequent complete response on anti-PD-1 therapy, clonotypes that arose during vaccination overlapped with T cell clonotypes found in the relapsed tumor and were persistent for nearly a year. While personalized vaccines have led to impressive long-term responses in some patients, and several clinical trials are currently underway, there is still work to be done to improve neoantigen vaccines. In addition to enhancing neoantigen prediction, optimizing timing of combination treatments, and lowering costs, Wu and her team are looking for ways to target new classes of neoantigens, including unannotated ORFs, splice variants, and gene fusions. They are also considering ways to increase HLA expression within tumors, in order to reveal hidden neoepitopes. Furthermore, in an ongoing clinical trial of renal cell carcinoma, NeoVax is being locally administered with subcutaneous anti-CTLA-4. The induction of robust circulating neoantigen-specific T cell responses shows early promise, and tissue samples are being taken to understand vaccine uptake by APCs in the skin. With numerous collaborative efforts underway in neoantigen vaccine research across Boston and around the globe, Wu expects to see significant progress in this field in the coming years.
Human CLEC9A antibodies deliver NY-ESO-1 antigen to CD141+ dendritic cells to activate naïve and memory NY-ESO-1-specific CD8+ T cells - Kristen Radford, PhD - Mater Research Institute
Monocyte-derived dendritic cells that are currently being used as cancer vaccines are safe, but produce low clinical response rates and are probably not optimal for inducing antitumor T cell responses. Kristen Radford focuses her work on creating off-the-shelf DC vaccines that target antigens to human CD141+ (BDCA-3+) DCs (equivalent to murine Batf3-dependent, CD103+ tissue-resident and CD8α+ lymphoid-resident DCs; cDC1). CD141+ DCs are rare in blood, but very effective at uptake and cross-presentation of cellular antigens, and at driving CD8+ T cell responses. The presence of CD141+ DCs in human cancers is associated with good prognosis and responses to anti-PD-1 checkpoint inhibition. Radford and her team have generated a human IgG4 antibody targeting CLEC9A, a molecule that plays an important role in the recognition and endocytosis of necrotic cells and in antigen cross-presentation, and is exclusively expressed on CD141+ DCs. Their CLEC9A antibody delivered human tumor antigens to CD141+ DCs for cross-presentation of multiple tumor-specific CD8+ T cell epitopes more effectively than an antibody targeting DEC-205 (which is broadly expressed on human DC subsets and other leukocytes)and in vitro reactivated memory, (cytotoxic T cells from melanoma patients that had been previously vaccinated with NY-ESO-1 protein). Furthermore, vaccination of humanized mice expressing a NY-ESO-1-specific TCR with CLEC9A-NY-ESO-1, poly I:C, and Flt3L led to priming of naive tumor-specific T cells in vivo and facilitated ex vivo expansion of NY-ESO-1-specific T cells with polyclonal effector function and capacity to lyse NY-ESO-1-expressing tumor cells. The CLEC9A-targeting antibody was superior in its ability to activate a CD8+ T cell response compared to an antibody targeting the DEC-205 receptor,.
NK cells and T cell therapies
T cell Therapy of Cancer - Helen E. Heslop, MD – Baylor College of Medicine
In her Keynote presentation, Helen Heslop discussed a number of advances in T cell therapy utilizing TCRs and CARs. T cell therapy has a number of advantages, including highly specific receptors for antigen targeting, good biodistribution, strong effector functions, and self-amplification. Heslop and colleagues have extensively investigated T cell therapies targeting Epstein Barr Virus (EBV) and other viruses as potential solutions to treating patients who develop viral infections or EBV lymphomas following a hematopoietic stem cell transplant. When given prophylactically, these donor-derived EBV-specific T cells (EB-VSTs) were highly effective in reducing the development of EBV lymphoma, and persisted for many years. While studying this form of T cell therapy, Heslop and colleagues greatly decreased manufacturing time, increased the range of viruses targeted, and developed and tested off-the-shelf options with encouraging responses. There is even current research into whether this strategy could be used against COVID-19. Following evidence that this strategy was safe and effective using T cells against viral antigens, researchers investigated whether this strategy could be used to target tumor-associated antigens (TAAs), and developed a strategy to target 5 lymphoma-associated antigens. When this strategy was tested in clinical trials, it was shown to be both safe and feasible, and responses against all 5 target antigens were observed, along with an increase in responses against TAAs that were not targeted, suggesting that epitope spreading had occurred. Switching gears towards CAR T cell therapy research, Heslop discussed ongoing research into the development of CD30-targeted CAR T cells. Early results in Hodgkin lymphoma patients showed that this strategy has limited toxicity and improves patient outcomes. When targeting B-cell lymphomas, the systemic loss of B cells that occurs is manageable; however, a similar loss of T cells or myeloid cells would be problematic, making hematological malignancies that arise from these cell types much harder to treat. In order to target T-cell lymphomas, Dr. Heslop and colleagues have developed CAR T cell therapies targeting CD5, to be used in the context of a transplant-enabling therapy. Early evidence has shown that this strategy is safe and shows evidence of activity, with dose-escalation studies still ongoing. Beyond hematological malignancies, Dr. Heslop also discussed various strategies to develop CAR T cell therapies that might be better equipped to overcome harsh tumor microenvironments. According to Dr. Heslop, VSTs are an attractive platform for the generation of ‘off-the-shelf’ allogeneic CAR T cells that may overcome the limitations of autologous products. Transducing VSTs with a CAR construct generates bispecific T cells that could be activated and costimulated by virus-infected cells and supported by virus-specific CD4+ T cells, while simultaneously targeting a TAA. Currently, Dr. Heslop and fellow researchers are investigating this with EBV-specific T cells transduced with CD30-targeting CARs.
NK Cells - Andreas Lundqvist, PhD – Karolinska Institutet
Adoptive NK cell therapy has been shown to be safe and has delivered promising, but so far limited clinical efficacy, especially in solid tumors. Andreas Lundqvist and his colleagues tackled the question of how to increase the persistence of active NK cells within the tumor microenvironment, which is often defined by oxidative stress and accumulation of reactive oxygen species (ROS). NK cell activity is attenuated by ROS. Lundqvist and his team found that in NSCLC, the tumor center contains a lower number of NK cells compared to the periphery, but that NK cells in the center are enriched in thiols. Cell surface thiols can be oxidized by external free radicals, and thus act as a protective shield and reduce local ROS levels. The team also found that IL-15 increases the expression of thiols on NK cells and that IL-15-primed NK cells are better at killing their targets in the presence of ROS than IL-2-primed NK cells. Mechanistically, IL-15 activated mTOR in NK cells, and mTOR suppressed the expression of TXNIP, resulting in sustained thioredoxin activity and density of thiol groups on the surface of NK cells, conferring resistance to oxidative stress. NK cells with high surface thiol expression also facilitated immune infiltration into tumors, turning ‘cold’ tumors ‘hot’ in ex vivo tumor explant studies.
Back to Top
by Maartje Wouters, Ed Fritsch, Lauren Hitchings, and Ute Burkhardt



