
Last week, the ACIR team attended the AACR Tumor Immunology and Immunotherapy Meeting 2022 in Boston, MA. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
T cell therapies
Eric Tran
Melody Smith
Robbie Majzner
Tumor microenvironment
Zev A. Wainberg
Garry P. Nolan
Zihai Li
Shannon J. Turley
Amy Moran
Myeloid cells
Jennifer L. Guerriero
Thomas F. Gajewski
Checkpoint therapy
Ryan J. Sullivan
Padmanee Sharma
Christian Blank
T cell therapies
TCR-gene therapy targeting mutant KRAS- Eric Tran - Providence Cancer Institute, Portland, Oregon.
Eric Tran discussed the potential of TCR-engineered adoptive T cell therapy, as well as the challenge of finding strong target antigens. Past studies have largely targeted shared self-antigens, cancer germline antigens, and viral antigens, and while some clinical efficacy has been observed, not all patients respond, and some experience high toxicities. Tran noted that driver mutations would make ideal target antigens, as they are essential to the cancer biology and are likely present in the majority of the cancer cells. In an effort to target such “hotspot” neoantigens, Tran and colleagues evaluated a TIL-treated patient and found that a high portion of the TILs were reactive to mutant KRAS G12D. In this patient, TIL therapy resolved 6 out of 7 lesions at about 9 months post infusion. The remaining lesion progressed, and upon resection, was found to have loss of heterozygosity (LOH) at chromosome 6, which encoded HLA-C*08:02. From this patient, researchers identified 4 TCRs capable of recognizing KRAS G12D mutations, and engineered T cells with these TCRs. In a clinical trial, two HLA-matched patients with PDAC were treated with T cells engineered with two of these KRAS G12D-specific TCRs. The first patient, CRI-3061, who had metastasis to the lung and liver, was treated with TCR-transduced T cells following lymphodepletion with cyclophosphamide and fludarabine. After infusion and high-dose IL-2, the patient presented with the expected IL-2 toxicities, along with unexpected symptoms of grade 2 ICANS, which correlated with high cytokine release that spiked on day 4 in the serum. The ICANS resolved without intervention, the transferred T cells expanded in the blood, and the patient showed signs of short-term tumor regression, with shrinking or stable lesions, reduced pain, and reduced markers of tumor cells. Unfortunately, this patient did not achieve a clinical response by RECIST, and despite additional treatment with PD-1 blockade and chemotherapy, the patient progressed and died of disease. The failure of this therapy could not be attributed to loss of mutant KRAS, loss of HLA, defects in the IFNγ pathway, or failure of the engineered T cells to infiltrate or persist; the mechanism of immune resistance in this patient is still unknown. The second patient, CRI-4483, who had metastasis to the lung, was treated with engineered TCR T cells using a similar regimen, but with several modifications: anti-IL6R was added to the preconditioning regimen (to prevent CRS/neurotoxicity), the cyclophosphamide dose was reduced, fludarabine was omitted, and T cells were manufactured in the presence of a cytokine cocktail of IL-2, IL-7, IL-21, and TGFβ, versus just IL-2. The incorporation of TGFβ was intended to polarize T cells towards a tissue-resident-like phenotype. Following treatment, this patient did not experience ICANS, and analysis showed that cytokine production spiked at day 1 in this patient, versus day 4 in the previous patient. This patient experienced a 72% partial response by RECIST at 6 months, and engineered T cells persisted long-term, though at lower levels in the blood compared to the first patient. Looking into why therapy was more successful in the second patient, the researchers found that the infusion product for this patient was consistent with an effector-like T cell population. Evaluating the role of the cytokine cocktail versus IL-2, the researchers found that T cells manufactured in the cocktail showed a more tissue-resident memory-like phenotype, preservation of markers of a less differentiated T cells, more effector cytokine production, and increased in vitro killing at low effector:target cell ratios compared to T cells manufactured with IL-2 alone. After about a year, patient CRI-4483 showed slow progression of a lung lesion, which was resected and is currently under investigation. This patient was also very recently re-treated with cocktail-stimulated TCR-engineered T cells (this time with only one of the TCRs) plus anti-PD-1. Interestingly, the patient experienced ICANS on this recent round of treatment. Going forward, Tran and colleagues are continuing to investigate why treatment efficacy varies between patients, and why the inclusion of TGFβ in the T cell manufacturing cocktail improves in vitro T cell function. They are also looking into novel synthetic receptors to boost T cell activity in the TME, as well as combination immunotherapies to enhance the efficacy of T cell therapies in solid cancers.
Elucidating the impact of the microbiome on CAR T cell therapy- Melody Smith - Stanford University, Stanford, California.
Melody Smith spoke on the importance of the intestinal microbiome in cancer immunotherapy, including CAR T cell therapy, where major advances have been made, but still a high portion of patients relapse or fail to respond to treatment, and many experience CAR-mediated toxicities. Curious about host extrinsic and intrinsic factors that might influence such outcomes, Smith turned to past studies that evaluated the influence of antibiotic exposure prior to CAR T cell therapy, and found that while prior exposure to antibiotics in general did not seem to affect survival in CAR T cell-treated mice, longer persistence of CAR T cells and prolonged B cell aplasia were observed in antibiotic-treated groups. In more recent clinical data, higher alpha diversity has been associated with favorable effector:target ratios. Also, the abundance of two taxa, Pediococcus and Anaerovoracaceae, were associated with improved CAR T cell expansion in vivo. Another group that looked at alpha diversity found that patients with high alpha diversity had increased survival and progression-free survival. Based on these past observations, Smith and collaborators hypothesized that the composition of the intestinal microbiome before CD19 CAR T cell therapy could be associated with clinical outcomes in patients with B-cell ALL or NHL. A cohort of 228 patients treated with any antibiotics within the four weeks prior to CAR T cell therapy showed a slightly lower overall survival upon treatment. However, many of the antibiotics that were evaluated in this cohort had a limited effect on the intestinal microbiome, so Smith decided to look more closely at stronger antibiotics that targeted obligate anaerobes, which were classified as P-I-M. This revealed that exposure to P-I-M had a much stronger effect in reducing progression-free and overall survival. Further evaluation confirmed that exposure to P-I-M was not merely a surrogate for sicker patients, as exposure to cefepime, a broad-spectrum antibiotic given to similarly sick patients, was not associated with worse progression-free or overall survival, while piperacillin/tazobactam exposure was. Further, other clinical factors, including ECOG and LDH, were both associated with reduced survival, but independently of P-I-M. In a fecal microbiome cohort, researchers evaluated baseline fecal samples prior to CAR T cell therapy for bacterial taxa and metabolic pathways to determine the association between the intestinal microbiome and CAR T cell efficacy. In 15 of 45 baseline fecal samples, “dominance”, defined as 30% or more of the microbiome consisting of a single taxon was observed, with the most frequent dominance being of the genus Akkermansia (6 of the 15 patients). Alpha diversity (diversity between taxa) at baseline was lower in patients compared to a healthy volunteer cohort, whereas the beta diversity (diversity within taxa) was much higher in patient samples, suggesting that patients with cancer are more likely to present with dysbiosis prior to therapy. Looking at biomarkers associated with day 100 complete responses, the researchers noted enrichment in microbial taxa within the class Clostridia, including the genera Ruminococcus and Faecalibacterium, and species Faecalibacterium prausnitzii. Notably, the abundance of these same microbes has also been associated with response in the context of immune checkpoint blockade therapy. In untargeted effect size analysis, Ruminococcus, Bacteroides, and Faecalibacterium were associated with day 100 complete responses, while Akkermansia was the top enriched dominant taxon in these patients. Smith also noted that while their research has focused on the taxa, their influence on outcomes may be more closely related to the metabolites they produce.
Engineering CAR T cells for enhanced efficacy and specificity- Robbie Majzner - Stanford University School Medicine, Stanford, California.
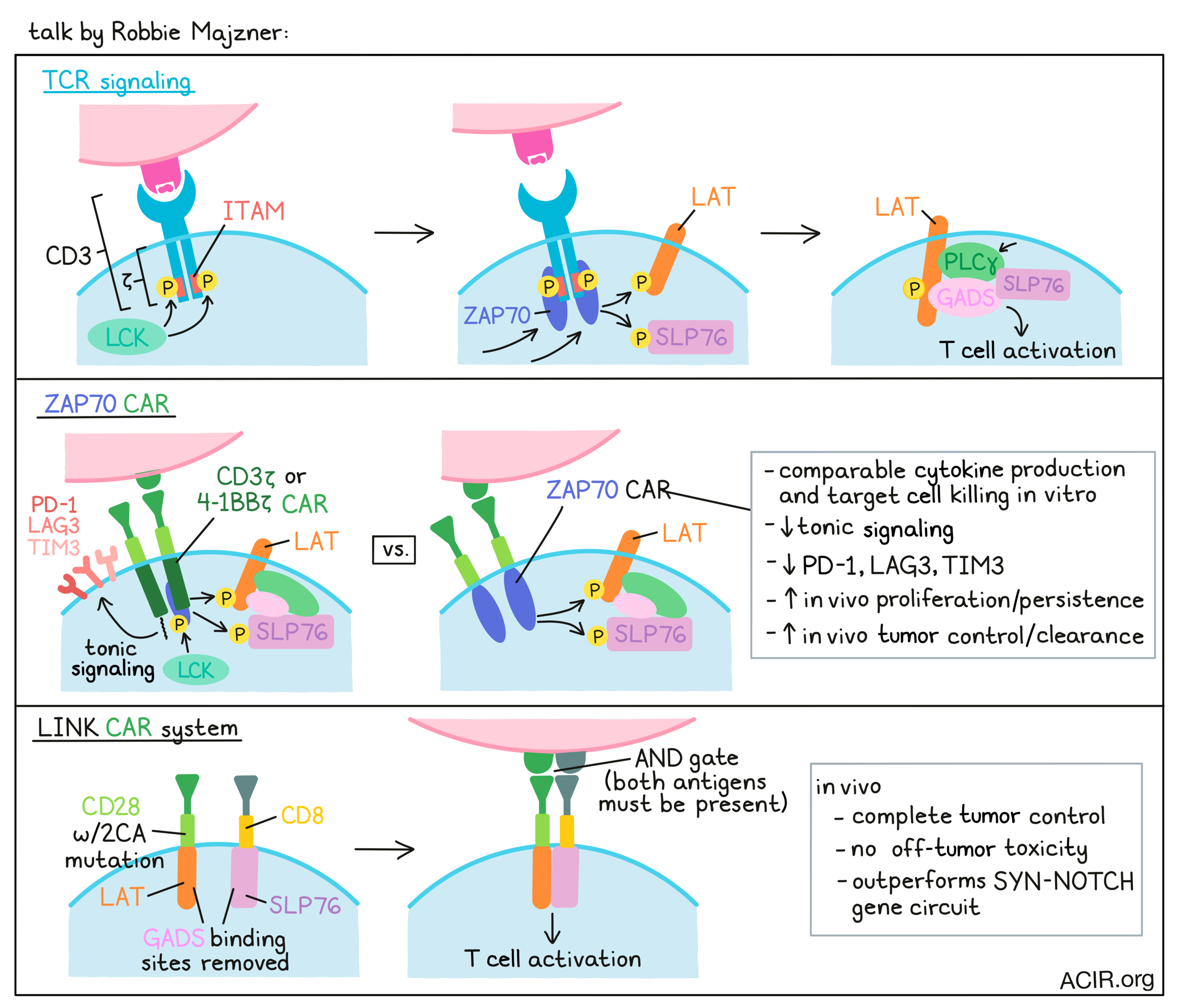
Robbie Majzner’s talk focused on a new approach to CAR T cell engineering. The vast majority of CAR T cell designs rely on CD3ζ, taking advantage, albeit crudely, of TCR signaling machinery to induce T cell activity. In TCR signaling, multiple ITAM-containing domains within the TCR, the most important of which are in CD3ζ, are phosphorylated by the kinase LCK, allowing for the ZAP70 kinase to come in and dock on the phosphorylated ITAMs, become activated, and go on to phosphorylate LAT and SLP76. These two molecules then come together to support the docking and activation of PLCγ, leading to multiple features of T cell activation. To show that CARs are dependent on the same proximal signaling networks, researchers at Majzner’s lab used CRISPR to knock out different proximal signaling molecules and found that LCK, ZAP70, LAT, and SLP76 were all necessary for CAR T cell activation. To determine what might be sufficient for CAR T cell activation, they replaced the CD3ζ chain and costimulatory regions with these proximal signaling molecules. Interestingly, the CAR that incorporated ZAP70 and the CAR that incorporated PLCγ had effective signaling activity and induced IL-2 production. The ZAP70 CAR, which expressed better than the PLCγ CAR, worked as well as the 4-1BBζ counterpart in terms of both cytokine production and target cell killing. However, the ZAP70 CARs showed reduced tonic signaling and induced fewer exhaustion markers, including Lag3, Tim3, and PD-1. In vivo, the ZAP70 CAR dramatically outperformed a 4-1BBζ CAR, inducing complete tumor clearance and cures in several models, likely due to enhanced proliferation and persistence. The activity of the CAR was independent of the endogenous TCR, but was dependent on the kinase domain for cytokine production and tumor killing. Further, it was not dependent on LCK, but was dependent on downstream LAT and SLP76, the two molecules needed for stimulation of PLCγ. In an effort to further bypass cellular mechanisms, the researchers next engineered individual CARs containing LAT or SLP76 on separate chains – each chain targeting a different antigen – with the idea that they could ligate together to propagate a signal only when both antigens were present. In a model targeting CD19 and HER2, CAR signaling did indeed drive IL-2 production in response to both antigens, but the initial design was “leaky”, allowing for responses in cases when only one antigen was present. Altering the hinge and transmembrane domain regions between constructs (CD28 and CD8) to prevent homodimerization, and introducing 2CA mutations into the CD28 transmembrane domain to prevent heterodimerization both helped to clean up the system, but some leakiness persisted. Next, the researchers tried interrupting binding of GADS, an adaptor molecule through which LAT and SLP76 interact, by removing the GASDS binding sites. Interestingly, rather than preventing LAT and SLP76 from interacting, interrupting GADS instead led to a completely clean system, with no response to single antigen-expressing cells, but robust killing of dual antigen-expressing targets. To test this specificity in vivo, they used ROR-1 as a model antigen with known on-target off-tumor toxicity in lung tissue. While the original mismatch system improved survival, the CAR T cells with the GADS mutation or GADS combined with 2CA allowed for complete tumor control without inducing toxicity. Stacking this new LINK (Logic-gated Intracellular Network) CAR system up against the SYN-NOTCH gene circuit, the current gold standard in the field, Majzner noted that their system acts as more of a true AND gate, rather than an IF/THEN system, and showed superior antitumor efficacy with less off-tumor toxicity results in vivo. The superior safety profile and true AND-gate status of this novel CAR T cell system could open the door for new antigens that could serve as safe targets for CAR T cell therapy.
Tumor microenvironment
Targeting the adenosine pathway to augment the immune response- Zev A. Wainberg - UCLA Medical Center, Santa Monica, California.
Gastric cancers have, to a large extent, not benefited from the first generation of immunotherapies, and so attention is now focused on combinations with the next generation of possible drugs. Zev Wainberg’s talk summarized approaches targeting the production and activity of the broadly immunosuppressive molecule adenosine. Extracellular adenosine is produced at an increased rate in actively growing tumors, as a product of enhanced consumption of ATP via two surface receptors: CD39, which converts ATP to AMP, and CD73, which converts AMP to adenosine. Adenosine appears to primarily function by binding to either of two receptors, A2AR and A2BR, which are found on a variety of immune cells, including dendritic cells and T cells. Despite early results suggesting a benefit to blocking interactions between adenosine and A2AR/BR, particularly with respect to myeloid cells, multiple trials of A2AR/BR inhibitors, anti-CD73, and (some) anti-CD39 have shown no single-agent activity, highlighting the need for combinations. Some encouraging results (improvements in ORR and PFS) have been seen with the anti-CD73 antibody oleclumab together with an anti-PD-L1 antibody (durvalumab) in non-small cell lung cancer (NSCLC). An early trial in locally advanced NSCLC following chemotherapy showed benefit of the combination over the standard follow-up therapy of durvalumab alone and intriguingly, may have been most beneficial in patients with low PD-L1 expression, therefore targeting a unique population. In a neoadjuvant study, oleclumab + durvalumab showed no improvement over durvalumab alone in terms of major pathological response rate, but patients with high CD73 expression may have benefited more, and the combination showed more T cells infiltrating the tumor. A large Phase III trial has been launched with a primary endpoint of overall survival. Turning to an anti-CD39 antibody, CTX030 (an IgG4 antibody) showed no toxicity in a phase I study, and doses were chosen for follow-up studies based on pharmacokinetic and preclinical data. In a study in metastatic gastric cancer combining CTX030 with chemotherapy and an anti-PD-1 antibody (budigalumab), the response rate was perhaps higher compared to prior studies without CTX030, but again there was the suggestion that patients with low PD-L1 expression may benefit more, particularly regarding durability, and could be a targeted population. Finally, early results from monitoring CD73 expression levels showed a tendency for CD73 expression to be higher in mutated KRAS-driven tumors, such as pancreatic, colorectal, and lung cancers. This was supported by TCGA data, suggesting that CD73 expression may be a negative prognostic marker. Studies in the KP KRAS-driven pancreatic model, looking at not just CD73, but also other enzymes that may impact adenosine levels, are currently ongoing, as are studies in early, borderline-resectable pancreatic cancer. In this setting, more tissue can be obtained for analysis and interventions may be attempted before the major immunosuppressive mechanisms set in with metastatic disease.
Cancer rearranges the rules in tissue building blocks. A new class of targets for therapy?- Garry P. Nolan - Stanford University School of Medicine, Stanford, California.
With a focus on understanding the architecture and phenotype of cell arrangements within tumors, Garry Nolan applied his multi-dimensional CODEX2 approach, which uses 50-120 oligonucleotide-labeled marker antibodies to identify tumor and immune cells in fresh frozen or FFPE tissue. Using a robotics platform for staining and fluorescence detection, along with advanced bioinformatics, this approach creates a wealth of information to look in and around cell “neighborhoods”. Nolan first focused on what happens at boundary areas between cell types to understand how the phenotype of the cells changed when interacting in these “perturbation zones”. Multiple markers were found to be upregulated and downregulated in these zones, and given the thousands of interactions (“experiments”) analyzed, rules for how cells change in the presence of other cells could be inferred and used to construct and compare tissue structures. For example, individual lymph nodes and tonsils were examined, and although there were some key, perhaps primordial, rules that were similar among all such lymphatic tissues, a number of differences were found, likely reflecting tissue specificity. Nolan speculated that cancers may be attracted to or excluded from certain lymphatic structures due to unique permissive or non-permissive rule sets. Analyzing multiple (~17) stage III/IV colorectal cancer samples with a CLR (Crohn's-like lymphoid reaction) histological diagnosis, which is known to be associated with a good outcome, and a nearly equal number of samples with a diffuse immune infiltrate (DII), which is associated with worse outcomes, he found that although the same neighborhood rules apply in both cases, the neighborhoods were much smaller and more fragmented in the DII samples. Focusing on CD4+PD-1+ cells, which are known to be predictive of outcome, Nolan found that the location of such cells was predictive only when in association with granulocytes. Dynamic changes, such as antigen activation, could be observed, and initiation, termination, and dysregulation events could be inferred in these tissue samples. Mapping neighborhoods for multiple cancer types and in different tissues, which is ongoing, is likely to reveal interesting new biological interactions, and may identify predictive markers. For example, although the presence of tertiary lymphoid structures is a good indicator of response, the presence of nearby T cells is a stronger predictor. Moreover, although a set of interactions between multiple cells (such as a triplet) may be similar in both CLR and DII samples, it is the interaction with a fourth cell (either a tumor or a germinal center cell) that differentiates the outcome. To allow experimental manipulation, Nolan described the use of pmel (gp100 transgenic TCR) cells in a murine melanoma model and bioinformatically constructed a map of all TCR-T cell interactions with other cells in the tumor over time. In this model, he was able to dynamically map how the TCR-T cell engineers the tumor, showing a progression of interactions from tumor cells, to endothelial cells (presumably to build blood vessels), and then to DC and myeloid like cells as PD-L1 turns on. Finally, Nolan investigated the metabolic inhibitor 2-HC, which has been shown in prior work to induce “super” CD8+ T cells with a more dominant memory phenotype than conventionally activated T cells. In this model, 2-HC-activated TCR-T cells yielded a dramatic change in cell neighborhoods, with a significant increase in the size and breadth of distribution of “productive” interaction zones between tumor cells and TCR-T cells, while for canonically activated cells, such zones were more sparsely distributed. Overall, the rules and dynamics of interactions that can be observed with this approach can lead to new biological insights and suggest novel therapeutic strategies.
Stomal Evolution in Inflammation and Cancer- Shannon J. Turley - South San Francisco, California.
In her keynote lecture, Shannon Turley introduced fibroblasts as the prototype stromal cells, and highlighted the role of LRRC15+ cancer-associated fibroblasts (CAFs) in immunosuppression and therapy resistance in cancer. Transforming growth factor beta (TGFβ) is a key immune regulator that activates fibroblasts in the tumor microenvironment, TGFβ-activated fibroblasts are associated with reduced survival and lack of response to ICB in immune-excluded human bladder tumors. To understand the role of TGFβ-activated fibroblasts in lack of response to ICB, Turley’s team analyzed the stromal compartment of pancreatic ductal adenocarcinoma (PDAC) – a highly aggressive and treatment-refractory disease in which fibroblasts and TGFβ are implicated in tumor progression. Single-cell analysis of early- and late-stage tumors from KRasG12D-driven PDAC tumor model and of healthy pancreatic tissues identified two major CAF subtypes that arise from healthy tissue fibroblasts during PDAC progression. Either TGFβ or IL-1 were computationally inferred as the key driver in each of the two major CAF subtypes in PDAC, and the two types differed in particular phenotypic features (matrix deposition and cytokine production). In the late-stage tumors, TGFβ-driven CAFs outnumbered other CAFs and became the dominant CAF subtype. In humans, scRNAseq identified similar major CAF clusters, including a dominant TGFβ-activated subtype, highlighting that distinct and overlapping molecular programs evolve in CAFs during human PDAC progression also. TGFβ-activated CAFs expressed the unique marker LRRC15, which is highly expressed in mouse and human pancreatic tumors. LRRC15+ CAFs showed peritumoral localization along with T cells and were highly enriched in stroma of PDAC and other human cancers. A TGFβ-activated CAF signature was also enriched in multiple human cancers, and was associated with reduced survival in ICB trials. In silico lineage tracing predicted that Pi16+ fibroblasts could give rise to all subsets of fibroblast in healthy tissue, including LRRC15+ myofibroblasts in a perturbed state. Analysis of Pi16+ fibroblast in PDAC mouse tumors identified that dermatopontin (Dpt) marks the universal fibroblast that gives rise to activated LRRC15+ CAFs, confirming the developmental relationship inferred by in silico data. Using an in vivo genetic approach, Turley and colleagues further demonstrated that universal Dpt+ fibroblasts gave rise to LRRC15+ CAFs in a TGFBR2-dependent manner in PDAC. In subcutaneous and orthotopic tumor models, selective depletion of LRRC15+ CAFs led to enrichment of normal universal fibroblast-like activity and significantly delayed pancreatic tumor growth in a CD8+ T cell-dependent manner. Therapeutic depletion of LRRC15+ CAFs reduced CD8+ T cell exhaustion marker expression and augmented effector functionality, whereas LRRC15- CAFs were immune-supportive. LRRC15+ CAF depletion potentiated antitumor immunity in response to anti-PD-L1 checkpoint blockade in subcutaneous and orthotopic settings. In addition to cancer, LRRC15+ fibroblasts were found to emerge in multiple human inflammatory diseases, including pulmonary fibrosis, COVID19, ulcerative colitis, and rheumatoid arthritis. LRRC15+ fibroblasts are enriched in active fibrotic lesions of Idiopathic pulmonary fibrosis lungs and in sites of active fibrosis in human Crohn’s disease.
Sex bias and cancer immunotherapy: Time has come- Zihai Li - The Ohio State University, Pelotonia Institute for Immuno-Oncology, Columbus, Ohio.
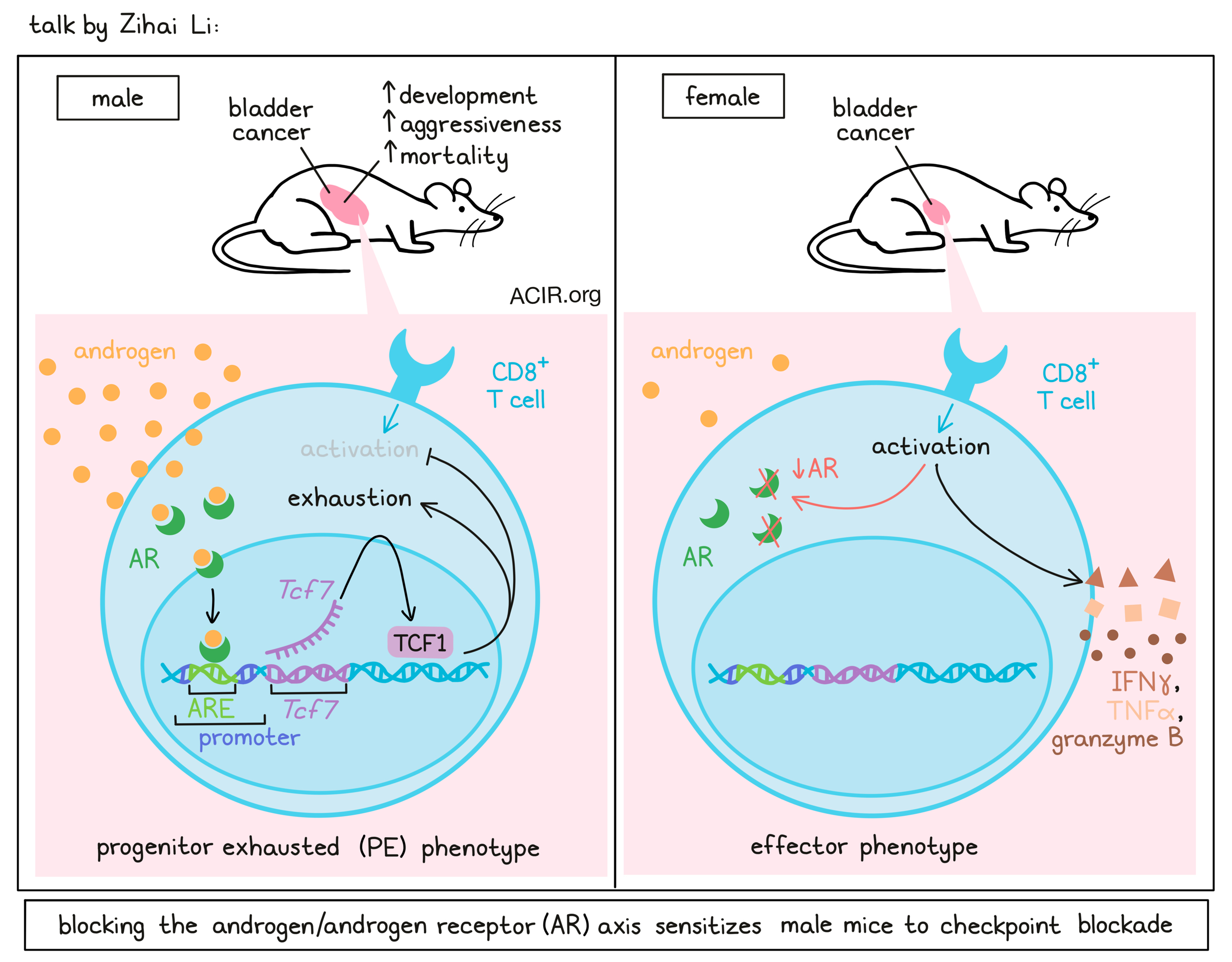
Zihai Li highlighted sex as an important biological determinant in cancer immunology and how sex bias suppresses T cell immunity and immune tolerance. Li defined sex as a biological determinant based on anatomy, sex chromosomes, sex hormones, and their interactions, as opposed to gender, which is a social and behavioral determinant. Sex chromosomes themselves can impact immunity, as several immune-related genes, such as TLR7/8, PDK, CXCR3, FOXP3, etc, are present in X chromosomes, which males only have only one copy of. Males also have higher mutational burdens and mortality across several cancers. For example, bladder cancer disproportionally impacts men, as the 4th most common cancer in men and 11th most common in women. To determine what drives sex bias in bladder cancer, Li and colleagues used common MB49 bladder cancer mice. Wild-type mice showed delayed tumor growth in female mice compared to male mice. However, the difference in tumor growth was completely abolished in Rag2-/- or TCR-knockout mice, but not in B cell-deficient mice, suggesting that endogenous T cell immunity drives the sex dimorphism in this tumor model. The same phenomenon was observed in several other cancers, where CD8+ T cells were both sufficient and necessary for sex bias. Single-cell analysis of CD8+ T cells at day 10, when the tumor size was not yet different, showed that distinct clusters for progenitor exhausted T cells were enriched in males, while effector-like clusters were enriched in females. The progenitor-like cluster in male mice was driven by Tcf7, Fos, Bcl2, and cJun, whereas there was female bias for genes involved in cell migration, effector function, and activation. In males, a Tcf7 transcription program drove the T cell exhaustion. To gain better mechanistic insight into this sex bias, Zihai and colleagues used sex-reversed four core genotype mice and saw that all male mice developed rapid tumors, dependent on the male hormone-associated gene Sry. This suggested a potential role of male hormones in regulating T cell immunity. Single-cell RNAseq data analysis of day 10 MB49 tumors showed that androgen-responsive genes were expressed in progenitor-exhausted CD8+ T cells; this was further confirmed across multiple datasets. Multiplex imaging further showed that AR was expressed in Tcf1+ (the gene product of the Tcf7 gene) progenitor exhausted T cells (TPE) and drove the Tcf7 regulon in TPE cells. Furthermore, reporter assays showed that AR activation transactivated Tcf7, and chip assays in AR-overexpressing Jurkat cells showed a 7-fold increase in AR binding to the Tcf7 promoter. To determine if androgen blockade could rejuvenate exhausted CD8+ T cells, MB49 tumors were grown in surgically castrated male mice. Castration reduced the tumor growth in male mice in CD8+ T cell-dependent manner. AR deletion in CD8+ T cells in both male and female mice abolished the sex bias in tumor growth; however, it did not enhance the antitumor efficacy of immune checkpoint blockade. High-dimensional flow analysis showed a dominant class of Tox+Tcf1- terminally exhausted T cells in wild-type mice, whereas AR-KO mice, anti-PD-1-treated mice, and AR-KO mice treated with anti-PD-1 all showed increases in effector T cells. In human patients, single-cell analysis of basal cell carcinoma (BCC) and non-small cell lung cancer (NSCLC) showed an increased T cell exhaustion program, suggesting the human relevance of sex bias. In a 30-year-long prospective study of 8771 men and women, high testosterone level was associated with a 30-80% increased risk of early death. In a pan-cancer setting, however, response to ICB showed heterogeneity in sex bias, highlighting the need for the consideration of other factors.
An unexpected role for androgens in limiting cancer immunotherapy efficacy- Amy Moran, Oregon Health & Science University, Portland, Oregon.
The main premise of Amy Moran’s talk was that the immune system is sexually dimorphic, shaped by sex chromosomes and hormones. This is well supported by deep epidemiological evidence of higher rates of autoimmunity in females, and higher rates of cancer and cancer mortality in males for a number of cancer types not associated with reproduction. Despite seemingly great targets in prostate cancer, immunotherapy has largely failed in the treatment of advanced prostate cancer in males, and the standard of care is blockade of the hypothalamus/pituitary axis through central hormone inhibitors, which drops levels of testosterone (chemical castration). Upon progression, treatment moves forward with the addition of small molecules that limit testosterone production via enzymatic activity, and/or small molecules that block binding to the androgen receptor (AR). The small molecule enzalutamide (ENZA) prevents androgen binding to AR, nuclear translocation of androgen-receptors, and AR binding to DNA. Given that T cells and IFNγ are essential in the control of tumor growth, Moran noted that T cells express AR in various tissues, with rapid upregulation of androgen receptors upon TCR ligation. Upon further investigation, they were able to show that androgen receptor engagement inhibits CD8+ T cell production of IFNγ. Looking at clinical responses to PD-1 checkpoint blockade in patients with advanced prostate cancer who were progressing on ENZA, Moran and colleagues found that patients had differential responses. Untangling the biology of this in an aged mouse model of castration- and immunotherapy-resistant advanced prostate cancer, in which anti-PD-1 alone or ENZA alone were ineffective, the combination of anti-PD-1 plus ENZA was shown to enhance survival. The T cells coming out of these tumors showed increased IFNγ and TNFα production. To determine whether this effect was T cell-intrinsic, Moran and colleagues used a TCR-transgenic mouse model (P14) of chronic LCMV infection. In this setting, transferred antigen-specific T cells with the AR CRISPR’d out were maintained at higher levels and resisted exhaustion, producing IFNγ at higher levels than their wild-type counterparts. When mice treated with wild-type T cells were started on ENZA halfway through infection, reflecting a more realistic disease treatment setting, an intermediate result was observed. A strong negative correlation between AR activity and IFNγ was observed in a number of clinical datasets, including in a set of CAR T cell-treated patients. Interestingly, this correlation was observed in both males and females. Next, Moran discussed the potential role of AR in tumor cells, and noted that prostate cancer has particularly low MHC-I expression. Investigating factors influencing tumor MHC-I expression, Moran and colleagues performed a whole-genome CRISPRi screen and identified AR as a dominant repressor of MHC-I expression in prostate cancer cells. In a prostate cancer cell line that does not express AR, MHC-I was naturally high, but forced AR overexpression reduced it, while androgen receptor knockout increased MHC-I, particularly in the presence of IFNγ. In mice, AR knockout in just tumor cells increased T cells in tumors. Piecing together an AR/IFNγ/antigen presentation axis, Moran used TCGA data to show that increased AR activity correlated with reduced IFNγ pathway activity and with reduced antigen processing and presentation. Together, these results suggest that androgen receptor inhibition can potentially enhance immunotherapy, both by enhancing T cell functionality and by making tumor cells more targetable. They also support the notion that hormones contribute to sex bias in cancer, and should be more thoroughly considered for their contributions to differential responses to immunotherapy.
Myeloid cells
The complexities of tumor associated macrophages and the key of effective targeting for anti-cancer therapy- Jennifer L. Guerriero - Dana-Farber Cancer Institute, Boston, Massachusetts.
Jennifer Guerriero described how macrophages are heterogeneous and highly abundant in a majority of solid tumors, and a high density of macrophages is associated with poor prognosis and resistance to chemotherapy. During tumorigenesis, tumor cell-derived factors recruit monocytes into the tumors, where they differentiate into tumor-associated macrophages (TAMs). These TAMs secrete chemokines and cytokines to promote tumor cell survival, angiogenesis, and metastasis; suppress antigen presentation machinery; recruit regulatory T cells; and inhibit antitumor CD8+ T cells. Several strategies are currently under investigation to target TAMs, including depletion or inhibition of pro-tumor TAMs and reprogramming of pro-tumor TAMs to an antitumor phenotype. BRCA1-associated triple-negative breast cancers (TNBC) have higher levels of cytosolic DNA and an active STING pathway, and are highly infiltrated with macrophages and T cells. The synthetically lethal PARP inhibitors Olaparib and Talazoparib are approved for the treatment of BRCA-deficient TNBC, and show better response compared to chemotherapy, but resistance ultimately emerges, blunting the long-term survival. In a BRCA1- and P53-deficient TNBC cancer model, which recapitulates the clinical Olaparib resistance phenotype seen in clinical trials, PARP inhibition efficacy was dependent on cGAS/STING pathway activation and CD8+ T cell recruitment. This suggested the combination of PARP inhibitors with ICB as a potential therapeutic combination in TNBC. However, clinical trials combining PARP inhibitors with ICB failed to demonstrate superior activity compared to PARP inhibitor monotherapy. To check if the failure of ICB in this TNBC clinical trial was due to the high number of macrophages in the TNBC TME, Guerriero and colleagues analyzed macrophages in BRCA/P53-deficient GEMM after PARP inhibitor treatment and found increases in suppressive tumor macrophages. PARP inhibition directly modulated human macrophages, leading to differentiation of CD14+ monocytes into macrophages with increased expression of CSF-1R ex vivo. In a BRCA/P53-deficient tumor model, anti-CSF-1R enhanced PARP inhibitor efficacy in a CD8+ T cell-dependent manner and overcame resistance to PARP inhibition. Mechanistically, PARP inhibitor-induced macrophages suppressed T cell function and induced T cell apoptosis via secreted factors. Single-cell RNAseq and proteomic analysis of PARP inhibitor-treated macrophages revealed that these macrophages were dependent on lipid metabolism. In particular, the SREBP1-mediated fatty acid synthesis pathway was shown to be associated with pro-tumor macrophages. Fatostatin, a SREBP1 inhibitor, in combination with Olaparib and anti-CSF-1R led to complete tumor elimination in the BRCA/P53-deficient tumor model. Single-cell analysis of human TNBC patient samples showed BRCA1-associated tumor cells and macrophages both had increased lipid metabolic signaling, which was further enhanced by PARPi, suggesting the potential for combining metabolic perturbation with PARP inhibitors to dampen the immunosuppressive effects of macrophages.
Commensal microbiota and germline variants can regulate anti-tumor immunity through myeloid cells- Thomas F. Gajewski, University of Chicago.
Thomas Gajewski’s presentation began by distinguishing T cell-inflamed and non-inflamed tumors, which differ in CD8+ T cell infiltration, chemokines, type I IFN gene signatures, and more. Under these differential immune pressures, tumors utilize different mechanisms of immune escape, with non-inflamed tumors escaping via immune exclusion, and inflamed tumors escaping via antigen loss or utilization of immune-inhibitory pathways. Compared to non-responders, most patients who respond to immunotherapy have inflamed tumors and an activated T cell signature at baseline. Curious as to what factors might contribute to these differences between patients, Gajewski and colleagues investigated potential sources of inter-patient heterogeneity of the T cell-inflamed tumor microenvironment, with consideration towards tumor cell somatic differences, environmental differences, and host germline genetic differences in immune regulatory genes. Looking first at tumor cell somatic differences, Gajewski evaluated DNA repair machinery, Wnt/β-catenin activation, and PTEN loss. In regards to DNA repair machinery, Gajewski highlighted a paper that showed that overexpression of DNA repair machinery results in low T cell infiltration, but does not reduce neoantigens, as it shuts down the endogenous STING pathway. Studying Wnt/β-catenin signaling, Gajewski and colleagues found that active β-catenin signaling in tumors hindered the recruitment of Batf3 DCs, which mediated a non-inflamed phenotype and resistance to various immunotherapies. Looking more closely at at Batf3-lineage DCs, which are known to be involved in the initial priming of T cells and in the recruitment of activated T cells to the tumor, Gajewski and colleagues found that Batf3 DCs were also required in the tumor microenvironment to provide 4-1BB costimulation to T cells, and that without Batf3 DCs in this role, the efficacy of PD-L1 blockade was lost. In human tumors, they identified non-random aggregation of Batf3 DCs and CD8+ T cells. Next looking at environmental factors, Gajewski noted that the composition of the gut microbiome is associated with the efficacy of PD-1 blockade in metastatic melanoma. To better study this mechanism, Gajewski and colleagues developed reverse-translational mouse models, in which germ-free mice that had been reconstituted with microbiomes from responder and non-responder patients were bred, yielding progeny that stably maintained the parental gut microbiomes. FMT back forth between mice with responder and non-responder microbiomes revealed that non-responder microbiome mice could benefit from transfer of gut microbes from responder microbiome mice, and similarly, responder microbiome mice could be impaired by the transfer of gut microbes from non-responder microbiome mice. These results suggested the presence of both good and bad microbes that could influence outcomes. Delving into the underlying mechanisms, the researchers found that in the presence of a favorable microbiome, the macrophage population in the tumor microenvironment was enriched for M1-like genes, versus the M2-like genes observed in tumors in mice with an unfavorable microbiome. In the granulocyte compartment, responder microbiome mice were enriched for neutrophil-like genes, while non-responder microbiome mice were enriched for MDSC-like gene expression. Further, an M1:M2 ratio signature derived from this mouse data showed that patients who were non-responders to PD-1 blockade had a low M1:M2 ratio, suggesting that the patterns identified in mice may be consistent in patients. Finally, looking at host germline genetic differences, Gajewski showed that in TCGA data, host germline variants/polymorphisms of the PRKCD gene, encoding PKCδ (expressed in all myeloid cells), were associated with reduced PRKCD expression and an increased immune gene signature. Knockout of PKCδ in the bone marrow of mice was found to improve tumor control, and further investigation revealed that while priming and early recruitment remained unchanged in PKCδ knockouts, there was an increase in activated T cell accumulation over time, a shift from an M2-like to an M1-like macrophage phenotype, and a beneficial change in neutrophil subsets. In human datasets, a PKCδ-knockout gene signature was associated with clinical benefit in patients treated with single-agent PD-1 checkpoint blockade. Given that PKCδ is a kinase, it should be druggable with small molecules, which could allow for interventions that support a more favorable TME.
Checkpoint therapy
Uncoupling anti-tumor immunity and auto-inflammatory toxicity from immune checkpoint inhibition- Ryan J. Sullivan, Dana-Farber Cancer Institute, Boston, Massachusetts.
Ryan Sullivan presented his thoughts on how to uncouple antitumor immunity from immune toxicity following immunotherapy, and began by discussing a case study of a melanoma patient who developed a late brain recurrence, presumably from an initial skin lesion 10 years earlier. This patient was treated with ipilimumab/nivolumab (the preferred treatment for melanoma brain metastases), however, the second dose induced significant lung pneumonitis, which initiated a downward spiral of other inflammatory complications, despite multiple treatments. The patient ultimately died, and autopsy showed widespread aspergillus infection in the lungs and gut as the cause of death, with no evidence of melanoma in the lung or brain, thus dying of an opportunistic infection. Opportunistic infections are not included in typical databases of immune-mediated causes of death and thus may be underrecognized. The story of this patient eventually led to a more holistic approach to care for patients with irAEs. Aptly, Sullivan likened immune therapy to an inflammatory fire – we want to cook with it, but not burn down the house. He then posed this as the central challenge to immunotherapy: being able to capitalize on antigen-specific antitumor responses while managing the drugs used to reverse toxicity and prevent an anti-patient response. Vitiligo is an AE that is directly related to antitumor responses through the antitumor T cells in the skin, with lots of supporting evidence. However, a proven relationship to shared antitumor antigens and other sites of irAEs in other diseases is still lacking. Interestingly, irPneumonitis appears more common in diseases associated with chronic lung injury and tobacco use, suggesting criticality of other factors. irPneumonitis is sometimes proximally associated with tumors and may be a consequence of antitumor activity, further suggesting a different treatment strategy is warranted in some circumstances. Data in a high-dose IL-2 trial also led to the conclusion that steroid treatment eliminated treatment efficacy. In patients with melanoma treated with ipilimumab, the presence of irHypophysitis was associated with better outcomes, and treatment with high-dose steroids for the irHypophysitis mitigated outcomes. Multiple studies have shown an association between irAEs and benefit (overall response rate or relapse-free survival), and so the challenge is how to use steroids in the right way. One approach is diagnostic testing. For example, scoping patients on checkpoint inhibitors with diarrhea and suspected colitis revealed that ~30% did not have colitis, 30% had only microscopic colitis which could be treated effectively with budesonide and continued on ICB, and more aggressive treatment was only required for the remainder of patients, most of whom were removed from ICB therapy. Another approach may be more aggressive tapering (kidney nephritis as an example). Finally, patients with life-threatening myocarditis appear to benefit with early high-dose corticosteroids. Trials targeting particular immune modulators such as TNF or IL-6, rather than using broadly active immunosuppressants such as corticosteroids, are underway as novel attempts to balance benefit and toxicity.
From the clinic to the lab: Investigating mechanisms of response and resistance to immune checkpoint therapy- Padmanee Sharma, MD Anderson Cancer Center, Houston, Texas.
Padmanee Sharma highlighted how neoadjuvant (pre-surgical) clinical trials support our understanding of the immune system and add to patient care. Neoadjuvant clinical trials allow us to move into earlier stages of disease and obtain sufficient samples, which can provide insights into clinical signals, biomarkers, and mechanisms of action. For example, the first neoadjuvant clinical trial of anti-CTLA-4 in patients with bladder cancer (n=12), done in 2006 (prior to FDA approval), provided important safety data and signals of clinical efficacy. Further, tissues collected at the time of surgery allowed for immune monitoring studies that helped to identify mechanisms of response and resistance. In this anti-CTLA-4 trial, T and B cell populations increased post treatment in the tumors, and ICOS pathway-related genes were the top differentially expressed genes. In another neoadjuvant clinical trial in patients with localized bladder cancer, the combination of anti-PD-L1 and anti-CTLA-4 led to 37.5% pathological complete responses (pCR). Samples obtained from surgical resections revealed that the responders in the trial had tertiary lymphoid structures with increased ICOS-expressing CD4+ T cells. Interrogating the role of ICOS in mice, Sharma and colleagues showed that ICOS/ICOSL KO mice demonstrated impaired tumor control with anti-CTLA-4 therapy, whereas targeting ICOS plus anti-CTLA-4 improved antitumor responses against B16 melanoma. Highlighting the utility of neoadjuvant therapy across different cancers, Sharma discussed a number of neoadjuvant trials that have yielded promising results. In a trial in lung cancer, anti-PD-1 and anti-CTLA-4 led to a number of major pathological responses (mPR). In the NeoCOAST trial, PD-L1- patients showed pathological responses following treatment with anti-CD73 in combination with anti-PD-L1. In a trial in cutaneous squamous cell carcinoma, anti-PD-1 neoadjuvant therapy led to the pathological responses. In yet another trial, neoadjuvant and adjuvant immune checkpoint blockade was evaluated in patients with high-risk, resectable metastatic melanoma. Here, anti-PD-1 monotherapy showed a pCR rate of 25%, while the combination of anti-PD-1 and anti-CTLA-4 showed a pCR rate of 45%, showing the potential benefit of combination therapy. They also identified characteristics that could be used to match patients with the treatments best suited for them. In another study in patients with melanoma, neoadjuvant anti-PD-1 therapy showed better PR, CR, and event-free survival compared to adjuvant therapy. Investigating “cold” versus “hot” tumor microenvironments, Sharma and colleagues performed a neoadjuvant therapy trial in 2009 in prostate cancer, which showed that anti-CTLA-4 converted cold tumors into hot, with increased immune cell infiltration. However, compensatory inhibitory pathways, such as VISTA and PD-L1 (expressed in CD68+ macrophages with mutual exclusivity) were upregulated in these tumors as well. Another observation in this study was that patients with bone metastases were less responsive to immune checkpoint blockade than patients with soft tissue metastases. Investigating this, Sharma and others found that soft tissue metastases show increased Th1 and INFγ responses, while bone metastases show increased Th17 responses. The researchers then replicated these results in a reverse translation mouse model, which they used to identify two cytokines, IL-6 and TGFβ, as drivers of the Th17 response in bone microenvironments. In mice with bone tumors, the combination of anti-CTLA-4 and anti-TGFβ led to an increase in Th1 CD4+ T cells, clonal expansion of CD8+ T cells, tumor regression, and improved survival. Finally, Sharma described a pilot safety trial in stage 4 metastatic clear cell renal cell carcinoma (ccRCC) in which immune checkpoint blockade prior to debulking surgery of a larger lesion (or lesions) provided good safety data. An ad hoc analysis suggested a potential clinical benefit, and tumor tissues from these patients showed that a higher IFNγ signature was associated with partial response at the population level, but not at the individual level. Tumors from patients with high IFNγ signatures, both responders and non-responders, showed that while a high IFNγ signature alone may not be sufficient to predict response, spatial interactions between T cell, B cells, and myeloid cells could predict better outcomes. Together, these results support the use of neoadjuvant clinical trials both to treat patients and to investigate emerging therapies and combinations.
Personalization of neoadjuvant therapy in melanoma - a template for other tumors?- Christian Blank - Netherlands Cancer Institute.
As explained by Christian Blank, neoadjuvant immunotherapy aims to utilize checkpoint blockade while significant tumor tissue is still present to provide a greater antigen source for T cell expansion than the limited micrometastases present after surgical removal of the tumor. This approach has been demonstrated in murine models, which showed that survival was dependent on NK, CD4+ T, and CD8+ T cells and on IFNγ signaling and perforin production. In a human trial, neoadjuvant checkpoint blockade led to a broader and stronger presence of tumor-specific T cells and a strong improvement in relapse- or event-free survival. An ongoing phase III trial is evaluating two doses of the ipilimumab/nivolumab combination prior to surgical resection, followed by a two-arm strategy after resection depending on the observed pathological change. Longer-term (5 year) follow-up showed strong overall (90%) and relapse-free (70%) survival, which compares very favorably to the expected <50% OS with surgery alone. Three-year follow-up in the OPACIN-neo trial showed similar high overall survival (>90%). A major (mPR) or a partial pathological response (pPR) at 6 weeks (after ipi/nivo neoadjuvant therapy) is a strong predictor of good overall survival, with weak responses indicating poor outcomes. Personalization of neoadjuvant immunotherapy can occur at multiple time points prior to, during, or after therapy, including looking for baseline markers prior to treatment, making on-treatment therapy adjustments based on pathological analysis of the resected tumor, or performing risk-adapted follow-up. As an example, in the PRADO trial (Personalized Response-driven Adjuvant therapy after Combination), the pathological response in the index lymph node (the largest node by radiological examination, which is then marked with a metal clip) determines whether or not a total lymph node dissection (TLND) is performed, and influences post-TLND therapy plan. These plans were developed based on retrospective analysis of the OPACIN-neo trial, which indicated a very strong correlation between the index lymph node response and the response of the entire set of lymph nodes. Surgically related morbidities were all reduced, and quality of life markers were improved when TLND was not required. At 2 years, patients with a major pathological response in the index node, who therefore had no further therapy, showed over 90% relapse-free survival. Early analysis of the partial pathological responders, who received TLND, but no adjuvant therapy, showed relapse-free survival that was worse than expected, leading to adjustment of the trial to include adjuvant therapy for this group also. Blank also described that patients showing a mPR in the index lymph node shifted to a follow-up without routine CT scans (which reduces patient stress), but with digitally followed patient-reported symptoms to identify potentially important events. Finally, application of measurement of an IFNγ-related transcription signature in the tumor at initial biopsy (baseline) allowed discrimination of which patients should receive nivolumab alone vs the combination of ipilimumab and nivolumab. Some patients still do not respond to the ipilimumab/nivolumab combination treatment, and a BATF3 signature, a marker of ability to cross-present antigen by dendritic cells, was found to be low in these patients. Blank then described assays to identify compounds that enhance cross-presentation, as well as the use of IL-2 to improve anti-PD-1 responses, which was identified in the work of Daniella Thommen using patient-derived tumor fragments. This therapy is now built into a neoadjuvant trial in melanoma. Overall, good outcomes following neoadjuvant immunotherapy and further personalization of these therapies to fine-tune and optimize responses provides a good opportunity to improve outcomes and quality of life for patients with cancer.
By Lauren Hitchings, Shishir Pant, and Ed Fritsch



