Many non-small cell lung cancer (NSCLC) tumors do not respond to immune checkpoint blockade (ICB), even in the presence of CD8+ tumor-infiltrating lymphocytes (TILs). To explore the mechanisms behind this phenomenon, Horton et al. evaluated T cell phenotypes and functions in orthotopic lung and flank mouse models for NSCLC. Their results were recently published in Science Immunology.
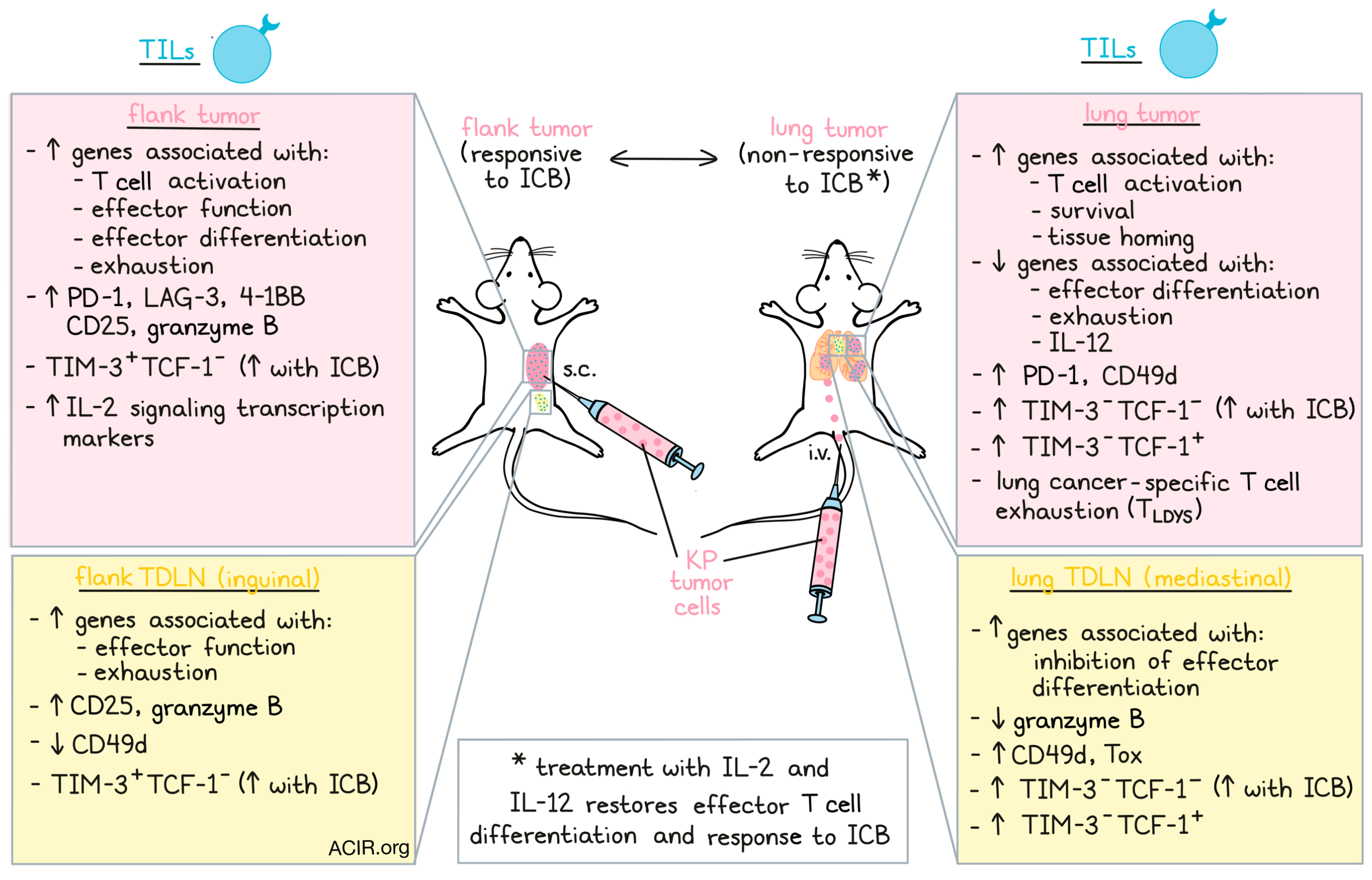
The model was created by injection of KP tumor cells either intravenously, to establish lung tumors, or subcutaneously, to establish flank tumors. Mice were treated with anti-CTLA-4 and anti-PD-L1 (ICB) starting on day 7 after inoculation. Even though ICB treatment slowed down the growth of the flank tumors, the lung tumors did not respond. This was not due to a lack of TILs, as the lung tumors had a higher TIL density than the flank tumors that responded to therapy. However, the ICB-responding flank tumors had a significantly higher TIL expansion.
The researchers then performed single-cell RNA sequencing (scRNAseq) on the TIL of both tumor locations. Nine distinct T cell phenotypes were found, and two distinct non-naive CD8+ T cell clusters were dominant either in the flank or the lungs. The cluster in the flank had increased expression of T cell activation and exhaustion genes, while the cluster in the lungs had upregulation of genes associated with T cell activation, survival, and tissue homing. Flank and lung TILs both expressed PD-1, but flank TILs expressed higher levels of LAG-3, 4-1BB, CD25, and granzyme B (GzmB), while lung TILs expressed higher levels of CD49d. These data suggested that lung TILs present with signs of T cell activation, but lack effector differentiation and exhaustion characteristics; the phenotypic state of these TILs was named lung cancer-specific T cell dysfunction (TLDYS).
TIL differences between the tumor sites were not driven by differences in antigen specificities or TCR usage, as there was clonal expansion in both T cell populations. There were also shared CDR3 sequences across the flank and lung tumors, suggesting that both tumors had responses to a shared antigen. Horton et al. then engineered KP cells to express the model H-2K antigen SIYRYYGL (KP.SIY). SIY-pentamer-positive CD8+ TILs were sorted from both tumor sites and analyzed for transcriptional differences. Again, the flank CD8+ subset expressed higher levels of exhaustion markers, and the lung subset was enriched for lung-homing factors.
Tissue staining revealed that PD-1+CD8+ T cells were more abundant in lung tumors than in the adjacent lung tissue, suggesting there was no TIL infiltration barrier. When TILs sorted from the flank or the lung were reconstituted in Rag2-/- mice and mice were inoculated with KP tumors in the flank, the flank TILs slowed down tumor growth more than the lung TILs, suggesting TLDYS is a persistent T cell state that cannot be reversed by a different tumor environment.
To study whether the differences between the populations were induced during T cell priming, Horton et al. adoptively transferred naive 2C T cells (reactive to SIY) into mice with KP.SIY lung or flank tumors and assessed the 2C T cells primed in the tumor-draining lymph nodes (TDLNs). T cells primed in the flank TDLNs expressed high levels of effector function and exhaustion genes, while lung TDLN-primed cells had high levels of genes associated with inhibition of effector T cell differentiation and other genes observed in the orthotopic lung tumor T cells. Additionally, flow cytometry confirmed an increase in CD25 and GzmB, and a decrease in CD49d expression on T cells activated in flank TDLNs.
In the KP.SIY model, SIY-reactive CD8+ T cells were found in equal numbers in both tumor types, but the cells in the flank TDLNs expressed higher levels of CD25 and GzmB, while the cells in the lung TDLNs expressed higher levels of Tox and CD49d. Additionally, T cells in flank TDLNs and tumors upregulated TIM-3 and were negative for TCF-1, while those in the lung TDLNs and tumors were TIM-3-TCF1+ or TIM-3-TCF-1-. Given that ICB induces the differentiation of precursor-exhausted CD8+ T cells into TIM-3+TCF-1- effector T cells, the researchers assessed the effects of ICB on the phenotype of the populations in both tumor types. While the flank tumors indeed had an increased proportion of TIM-3+TCF-1-SIY-reactive CD8+ T cells, there was an increase in TIM-3-TCF-1-CD8+ TILs in the lungs. When 2C T cells were isolated from flank and lung TDLNs and transferred to RAG2-/- mice with flank KP.SYI tumors, the ones primed in the lung TDLN retained their TIM-3-TCF-1+ phenotype with decreased GzmB, whereas those primed in the flank TDLN increased TIM-3 and GzmB.
To assess whether these populations were also found in human NSCLC, the researchers mapped the TLDYS and flank T cell signatures onto scRNAseq data of patient cohorts. Less-differentiated human CD8+ T cell populations showed similarity with the TLDYS state. Most of the CD8+ TIL were enriched for the TLDYS signature, while only a fraction were enriched for the flank T cell signature. In individual patients, there was a large variety in the contribution of each phenotype, and some patients completely lacked the flank T cell phenotype.
Given that the TLDYS cells had lower expression of IL-12-related genes, and the flank TILs had enriched levels of IL-2 signaling transcription markers, the researchers assessed whether treatment with IL-2 and IL-12 could prevent the induction of TLDYS. Mice bearing KP.SYI lung tumors were transferred with 2C T cells and simultaneously received IL-2 and IL-12 treatment. This treatment increased CD25 and GzmB expression in the mediastinal (lung-draining) lymph node. When endogenous SIY-reactive TILs were assessed in the TDLN and tumor after treatment with the cytokines, there was a downregulation of TCF-1 in the TDLN and upregulation of TIM-3, CD25, GzmB, and 4-1BB in the tumor and TDLN. Cytokine and cytokine plus ICB treatment both induced tumor growth reduction, resulting in prolonged survival.
In conclusion, in both mice and humans, lung tumors induced a specific dysfunctional T cell phenotype that limited tumor control and response to ICB, which could be overcome by IL-2 and IL-12 treatment. This provides a mechanistic rationale to test this cytokine in combination with ICB in NSCLC to increase therapeutic efficacy.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Brendan Horton answered our questions.

What prompted you to tackle this research question?
Several clinical studies have shown that response rates to immune checkpoint blockade vary not only between cancer types, but also between tissue sites of tumor growth. In patients, this can mean that not all tumor lesions will respond to immune checkpoint blockade, and therapy-resistant tumors can continue to progress. How the immune system can respond differently to similar tumors growing in different tissues is not well understood, and immune checkpoint blockade resistance in non-responding tumors is a major barrier to improving immunotherapy efficacy. We were interested in uncovering mechanisms to explain how differences in immune responses and responses to immune checkpoint blockade are established against tumors growing in distinct anatomic locations.
What was the most surprising finding of this study for you?
Resistance to immune checkpoint blockade is often associated with an absence of CD8+ T cells in the tumor microenvironment, however, we found that immunotherapy-resistant lung tumors were actually heavily infiltrated by T cells. Tumor-specific CD8+ T cells responding to immunotherapy-resistant lung tumors were activated, proliferated, and trafficked to tumors, but differentiated into a dysfunctional state that had very little ability to kill tumor cells and appeared distinct from the well-described state of T cell exhaustion. Most surprisingly, differentiation into this dysfunctional state didn't occur in the tumor microenvironment, but actually was induced during T cell activation in the tumor-draining lymph node, indicating these T cells were likely destined from the very beginning of the immune response to be resistant to the effects of immune checkpoint blockade. This implies that the conditions during T cell activation are another level of regulation that can influence responses to immunotherapy.
What was the coolest thing you’ve learned (about) recently outside of work?
I've been learning more about Italian amaro, which are herbal, bittersweet liqueurs used as aperitifs or in cocktails. A local distillery here makes their own version of amaro, but uses hops, which are usually used in beer making, as one of the major bittering agents. The result is totally delicious, which just highlights for me how classic things can be rejuvenated with the right combination of traditional and new approaches.



