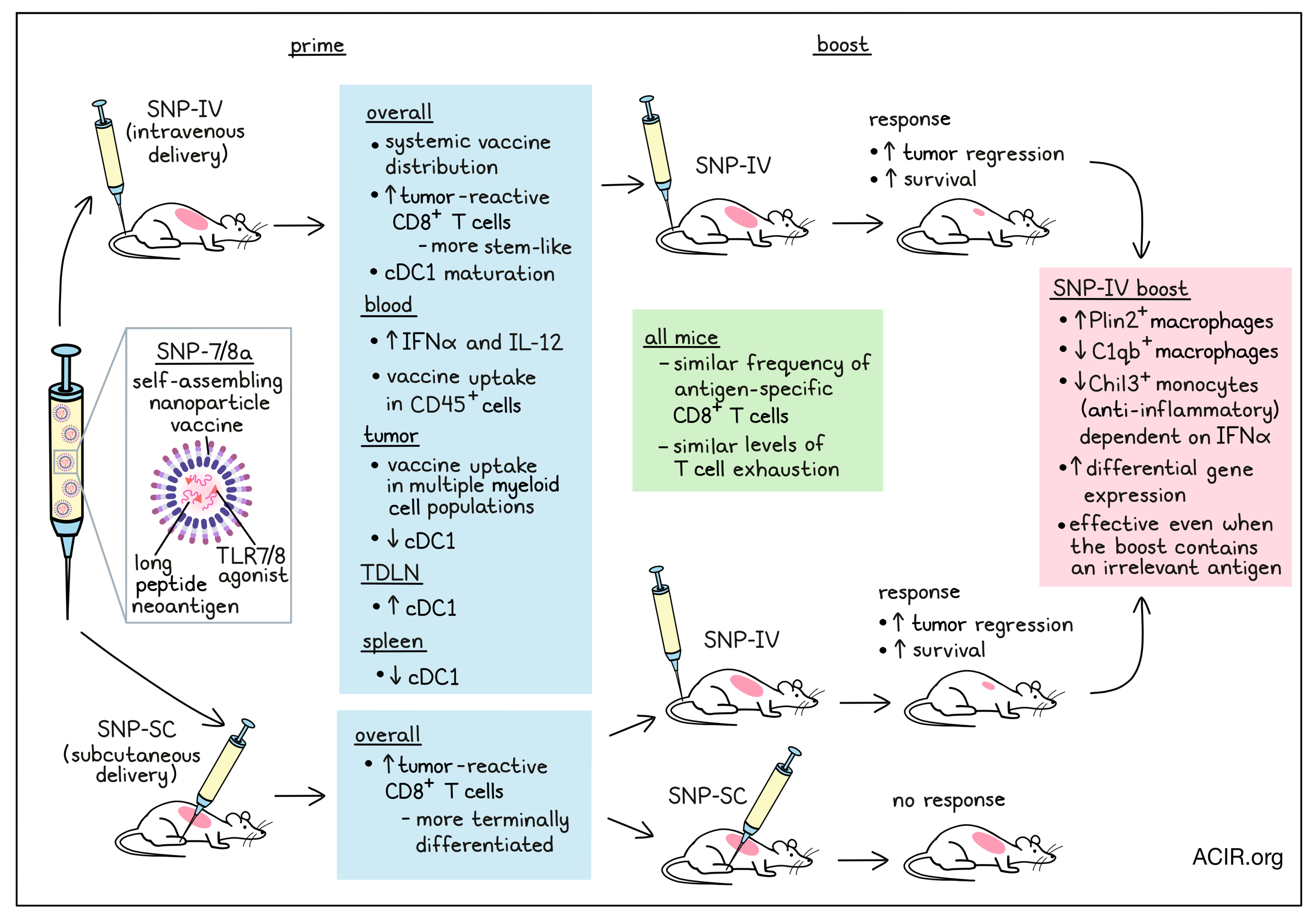
Since cytolytic T cells are critical to cancer immunotherapy, therapeutic cancer vaccines, which can induce such T cells, can be an important component of immunotherapy. However, current vaccine approaches to improve the magnitude of the immune response have traditionally shown limited antitumor effects. Taking a new approach, Baharom, Ramirez-Valdez, Khalilnezhad, et al. evaluated how the route of administration affects T cell responses and the tumor microenvironment (TME). Their results were recently published in Cell.
The researchers developed a self-assembling nanoparticle vaccine (SNP-7/8a) platform that delivers both long peptides containing neoantigens and a toll-like receptor 7/8 (TLR7/8a) agonist. Previously, they found that the route of administration of SNP-7/8a, either intravenous (SNP-IV) or subcutaneous (SNP-SC), altered the quality of generated neoantigen-specific CD8+ T cells, with SNP-SC generating mainly terminally differentiated CD8+ T cells, and SNP-IV generating mainly stem-like CD8+ T cells. To further assess the T cell response after vaccination, mice inoculated with MC38 tumors were treated on day 7 with a priming dose of SNP-7/8a containing the MC38 neoantigen Reps1, followed by a booster dose on day 14, together with anti-PD-L1. Tumors regressed and survival improved in the group primed and boosted with SNP-IV, while mice primed and boosted with SNP-SC did not experience tumor control. However, when mice were primed with SNP-SC and boosted with SNP-IV, tumor control was observed, similar to two doses of SNP-IV.
To confirm that neoantigen-specific CD8+ T cells were required to induce tumor regression, mice were vaccinated with SNP-IV containing an irrelevant antigen. This vaccine strategy did not induce tumor control, nor did SNP-SC priming with irrelevant antigen followed by SNP-IV with the MC38 antigen. However, priming with SNP-SC containing the antigen, followed by SNP-IV with an irrelevant antigen, did improve tumor control and survival. This suggests that the neoantigen-specific CD8+ T cells generated by SNP-SC can mediate tumor control when followed by systemic innate stimulation.
To further assess the T cell responses following SNP-SC or SNP-IV boost, the researchers collected blood on day 21 and measured tetramer+ T cells. SNP-SC twice or SNP-SC followed by SNP-IV resulted in similar frequencies of antigen-specific T cells, at ~10% of the total circulating CD8+ population. There was no difference in this response when an irrelevant antigen was used for the SNP-IV boost. Regardless of treatment, the tumor-infiltrating Reps1-specific CD8+ T cells had similar levels of the exhaustion markers PD-1, TIM-3, NKG2A, and CD39.
The researchers then assessed the vaccine pharmacokinetics and the differences between the two routes of administration. Systemic distribution of the vaccine was only detected after SNP-IV, not SNP-SC. Vaccine was detected in the spleen, tumor, and tumor-draining lymph nodes (TDLN) after vaccination with SNP-IV, not SNP-SC. A vaccine-positive CD45 population could be detected in the blood at six hours post-vaccination, and in the tumor, multiple myeloid cell populations took up the vaccine. Additionally, the serum of SNP-IV-vaccinated mice contained higher levels of IFNα and IL-12; given the role IFNα plays in promoting cross-presentation between cDC1 and CD8+ T cells, the cDC1s in the spleen, tumor, and TDLN were further assessed. Clustering revealed six clusters, with a reduction in cDC1 numbers in the spleen and tumor after SNP-IV, and an increase in cDC1 the TDLN. The cDC1s in the spleen, tumor, and TDLN following SNP-IV were also more mature (CD80+/CD86+).
To further assess the TME after vaccination, tumors and spleens were harvested 24 hours after boosting with the various vaccine regimens. Myeloid cells were sorted and subjected to scRNAseq. Untreated and SNP-SC-boosted mice had enrichment of Chil3+ monocytes in the tumor, while boosting with SNP-IV (relevant or irrelevant antigen) resulted in an enrichment of Plin2+ macrophages and a reduction of Chil3+ monocytes and C1qb+ macrophages.
Further assessment of the monocyte/macrophage compartment showed that more than 100 genes were upregulated after SNP-IV compared to 40 genes after SNP-SC. Furthermore, 300 and 55 genes were downregulated after SNP-IV and SNP-SC, respectively. Among the top differentially expressed genes (DEGs), Plin2+ macrophages upregulated genes associated with regulatory and suppressive function. These macrophages were also enriched in pathways related to adaptive immune responses, cell activation, TLR signaling, and regulation of TNF production. On the other hand, Chil3+ monocytes were enriched in pathways associated with anti-inflammatory responses. The researchers also found that genes associated with the coronavirus pathogenesis pathway, including genes related to IFN-I signaling and inflammasome activation, were upregulated. Conversely, genes associated with oxidative phosphorylation were downregulated after SNP-IV.
The researchers then assessed the role of IFN-I in the antitumor effects of SNP-IV. When mice were injected with blocking antibodies against IFNAR on days 13 and 15, IFNα was depleted, and the tumor reduction and survival effects of the SNP-IV boost were abrogated. However, the frequency of neoantigen-specific CD8+ T cells was not affected. Blockade of IFNAR reduced maturation marker expression on cDC1s and cDC2s in the spleen, tumor, and TDLN. When IFNAR was blocked, there was no depletion of Chil3+ monocytes after SNP-IV. Therefore, these data suggest that the effects of SNP-IV at the time of boost depend on IFN-I, which leads to depletion of the tumoral Chil3+ monocytes.
Finally, the researchers assessed whether these types of monocytes could also be detected in the human TME based on a list of human ortholog markers matching the top 50 DEGs in Chil3+ monocytes to interrogate an atlas of human monocytes/macrophages found in cancers. The huChil3 gene set was enriched in CD16- monocytes across various tumors. Bulk RNAseq samples from 364 tumors from 12 cancer types showed that the huChil3 gene set was enriched in myeloid cells, and from this, the huChil3+ monocyte abundance could be assessed. In pan-TCGA data, low huChil3 levels were associated with better survival, suggesting Chil3+ monocytes also play a negative role in the human antitumor response.
This novel research shows that modulating the TME following the induction of tumor-specific T cell responses can play an essential role in increasing the efficacy of cancer vaccines. It will be of interest to determine whether this vaccine strategy can also target the Chil3+ monocyte population in human tumors.
Write-up by Maartje Wouters, image by Lauren Hitchings.
Meet the researcher
This week, co-first author Faezzah Baharom answered our questions.

What was the most surprising finding of this study for you?
We were initially surprised that we did not need a boost (second dose of the vaccine containing antigen) to promote tumor regression. It turns out that low magnitude terminally differentiated tumor-specific CD8+ T cells that were generated by SNP-SC could still have cytotoxic activity if we overcome the suppressive tumor microenvironment. This could be a game changer in the clinic if we maximize the potential of tumor-specific T cells by reversing the inhibitory signals in the tumor instead of chasing higher magnitude T cells alone.
What is the outlook?
While the lab continues to explore the mechanism of how systemic innate immune activation improves the vaccine-mediated therapeutic response, another area of interest is to apply the “Vax-innate” concept to adoptive cell therapy (ACT). Here, we hope to improve the efficacy of ACT by providing systemic inflammation via i.v. injection of a vaccine/adjuvant after infusion of tumor-specific CD8+ T cells that were expanded ex vivo. Our collaborators are focused on advancing the vaccine platform to the clinic. I believe the future of cancer treatment lies in combination therapies, in which patients receive multiple treatment strategies simultaneously to improve their chances of defeating the disease.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently read about a golden age of Chinese philosophy, known as the Hundred Schools of Thought. It was a period of plasticity where philosophers experimented with different social concepts and moral codes. The idea that one could fine-tune moral values and challenge existing social norms intrigues and appeals to the scientist in me, especially in today’s society, where it seems like our views are more polarized and rigid.



