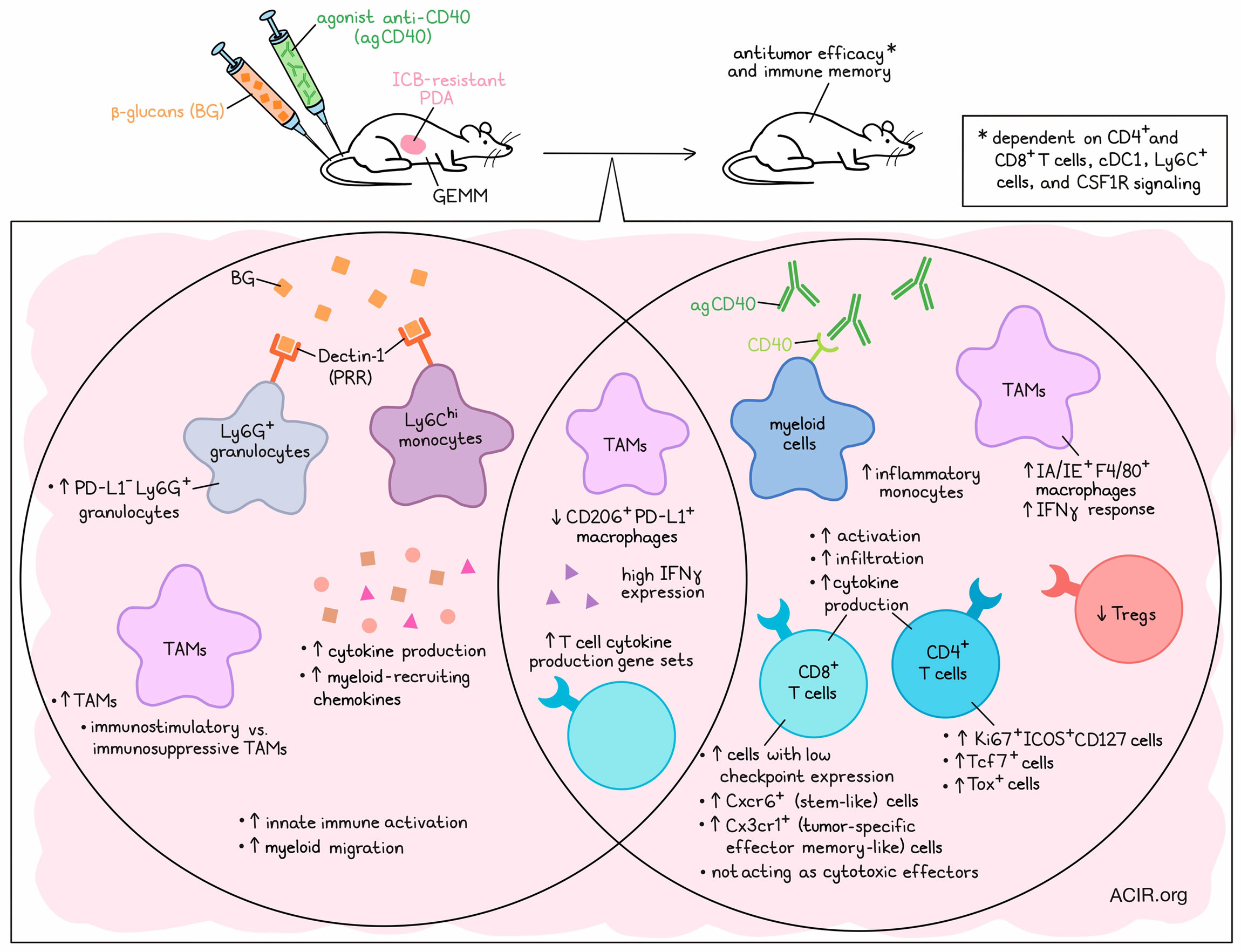
One of the reasons immune checkpoint blockade (ICB) may fail in solid tumors like pancreatic ductal adenocarcinoma (PDA) is a lack of T cell infiltration and extensive myeloid inflammation in the tumor microenvironment (TME). Wattenberg et al. aimed to overcome these characteristics in recent work published in Science Immunology.
Given the known plasticity of myeloid cells, the researchers started out by assessing publicly available scRNAseq data of human PDA to look for the expression of putative myeloid activating receptors. One of the most abundantly expressed TNF receptor family members was CD40. While strategies to target CD40 in early clinical studies have had limited effects, the researchers hypothesized that combining it with another agent targeting a myeloid signaling pathway might be more effective in triggering antitumor immune responses. The myeloid cells also expressed pattern recognition receptors (PRRs), frequently on clusters that lacked robust CD40 expression, suggesting these might be rational targets for combination therapy.
To test this hypothesis, the researchers made use of ICB-resistant GEMM PDA mouse models. As in the clinic, treatment with an agonistic CD40 antibody (agCD40) had a limited effect on tumor control. However, combining this therapy with any of several agonists of various PRRs resulted in high cure rates. The researchers focused subsequent work on targeting Dectin-1, which is a receptor for β-glucans (BG), because BG can be systemically administered in humans without causing significant toxicity. Combined treatment of BG and agCD40 in PDA models induced tumor control, without overt toxicity.
The researchers then performed mass cytometry (CyTOF) on orthotopic tumors to assess the underlying mechanisms of this synergy. A broad remodeling of the myeloid and lymphoid populations in the tumor was observed post-treatment. In the myeloid compartment, BG monotherapy increased the frequencies of PD-L1-Ly6G+ granulocytes, and a shift toward CD206loF4/80+ macrophages (CD206 is a marker of immunosuppressive macrophages) was observed. Meanwhile, agCD40 monotherapy increased inflammatory monocytes and IA/IE+F4/80+ macrophages. Combined treatment also induced these effects, and decreased immunosuppressive CD206+PD-L1+ macrophages. In the T cell compartment, agCD40 increased the frequency of Ki67+ICOS+CD127+CD4+ T cells and CD8+ T cells with low expression of immune checkpoints, while it decreased the frequency of Tregs. Bulk RNAseq of the treated tumors showed that BG resulted in the enrichment of genes related to innate immune activation, cytokine production, myeloid migration pathways, and myeloid-recruiting chemokine expression, while agCD40 was associated with increases in gene pathways related to lymphocyte and T cell activation.
To assess whether T cells were required for the effects of BG/agCD40, CD4+ and CD8+ T cells were depleted. Depleting both T cell subsets abolished the effects of therapy, and depleting one of the two populations resulted in a partial decrease in antitumor effects, indicating both populations were important. In cured mice, a second tumor challenge showed that treatment induced T cell-dependent immunological memory. Looking at the importance of other immune cell subsets, the researchers found that cDC1 were essential for the effects of the combined therapy, as in Batf3ko mice, deficient in cDC1, the antitumor effects were diminished. These data suggest that both T cells and cDC1 were required for the antitumor effects of BG/agCD40.
IHC analysis showed that agCD40 drove the increased tumoral infiltration of T cells. However, the infiltrated T cells in the TME after treatment were present surrounding the tumor lesions, not in the lesions. In mice lacking MHC class I and II or in Prf1ko mice, which lack perforin, the combination treatment remained effective. These data suggest that even though the T cells were required, they did not function as cytotoxic effectors in this setting.
Looking more in depth at the intratumoral T cells using scRNAseq data at 8 days after treatment, the researchers confirmed that the major changes in the lymphoid compartment were due to T cells and were driven by agCD40 treatment, with decreases in Tregs and increases in CD4+Tcf7+ and CD4+Tox+ clusters. There was also an increase in the frequency of CD8+Cxcr6+ T cells, a stem-like population, and CD8+Cx3cr1+ T cells, a tumor-specific effector memory-like population. Genes related to positive regulation of cytokine production were highly expressed in T cell subsets after agCD40 or combination therapy. Ifng was one of the highest expressed cytokines by T cells in combination treatment groups. Intratumoral IFNγ administration in the absence of agCD40 could also trigger the antitumor activity of BG therapy and increase survival in PDA models. These data suggest that agCD40 results in T cell infiltration and IFNγ production, which, combined with BG, mediates the antitumor effects.
Diving into the effects exerted by BG, the researchers assessed the Dectin-1-dependent binding of BG, and found that at 18 hours post-treatment, it mostly bound to Ly6Chi monocytes and Ly6G+ granulocytes, but not to TAMs, T cells, B cells, or CD45- cells. Tumors from treated mice showed an increase in the TAM-to-tumor cell ratios, suggesting that BG may remodel the TME and affect TAMs.
TAMs were common in the stromal compartment of the TME, and combination treatment increased intratumoral TAMs. scRNAseq data identified four TAM and two monocyte populations in the TME. CD206+ TAMs, previously associated with T cell exhaustion and protumoral characteristics, were found in the tumor border region, while CD206- TAMs were found in the intratumoral region. BG/aCD40 therapy resulted in a decrease in the ratio of CD206+/CD206- TAMs. TAMs showed increased immunostimulatory transcriptional programs, including defense response and biotic stimulus (stimulus from a component of a living organism), and CD206- TAMs had increased peroxisome activity after combination treatment. BG was a major driver of these changes, but upregulation of the IFNγ-responsive genes Stat1 and Ly6a was associated with agCD40 treatment, and pSTAT1tyr701+ TAMs could be detected in the TME, suggesting both treatments impacted the TAM populations.
Finally, to confirm the role of TAMs in treatment responses, BG/agCD40 treatment was repeated in combination with an anti-Ly6C antibody to deplete Ly6C+ cells, which showed reduced efficacy of the treatment. Additionally, pharmacologic targeting of CSF1-CSF1R signaling to inhibit systemic phagocytic cells and intratumoral TAMs also reduced efficacy.
In conclusion, this research shows the benefits of combining PRR stimulation (BG) and agCD40 treatment in preclinical ICB-resistant models of PDA, resulting in a profound remodeling of the TME and activation of immunostimulatory TAMs. This provides new avenues for clinical research in patients with PDA and other ICB-resistant cancer types.
Write-up by Maartje Wouters, image by Lauren Hitchings.
Meet the researcher
This week, first author Max M. Wattenberg answered our questions.

What was the most surprising finding?
One of the most surprising findings in our study was that cytotoxic T cells did not need to directly engage tumor cells for effective immunosurveillance. Rather, combinatory myeloid cell activating therapy drove T cells in the stroma to secrete immunostimulatory cytokines, which educated antitumor effector myeloid cells. These findings suggest new ways in which antigen-specific T cell responses might be leveraged to treat cancer. Indeed, our data show that by targeting myeloid cells, a concerted innate and adaptive immune response to cancer can be generated, bypassing classical mechanisms of immune evasion, including T cell exclusion and exhaustion.
What is next?
There are two key next steps for this work. First, we plan to define the safety, efficacy, and immunological mechanisms of combined myeloid activation in humans. To address this, we have initiated a Phase 1b clinical trial studying combined CD40 and Dectin-1 activation for the treatment of patients with pancreatic cancer (NCT05484011). Second, we aim to determine the molecular mechanisms by which myeloid activating therapy is mediating tumor cell killing, and in doing so, provide a roadmap for the development of next-generation myeloid-activating therapies.
What is the coolest thing you have learned outside of work recently?
I recently attended a college basketball game at The Palestra on the University of Pennsylvania’s campus, and was fortunate to see the Quakers upset nationally ranked Villanova. The Palestra, known as “The Cathedral of College Basketball”, opened in 1927, and hosted the East Regional of the NCAA’s first national tournament in 1939. Further, The Palestra has witnessed more college basketball games than any other arena, and is one of the first steel-and-concrete arenas in the United States.



