
Last week, the ACIR team attended the AACR Tumor Immunology and Immunotherapy conference in Boston, MA. This week’s extensive special feature is made possible thanks to the support of Takeda. It covers select sessions from the conference organized as follow:
Exhaustion and Therapy Resistance
Cell Therapy
Myeloid Compartment in the TME
Vaccines and Antigens
Microbiome
Exhaustion and Therapy Resistance
In one of two keynote lectures, Gordon Freeman from the Dana-Farber Cancer Institute described the history of how cancer immunotherapy came upon, unintentionally, a new strategy – stopping tumors from turning off the immune system. The initial observation that an antibody to CD28 stimulated T cells to proliferate and produce IL-2 triggered the hunt for the CD28 ligand, which was identified as B7.1 and, later, its stronger homologue B7.2 (aka CD80 and CD86, respectively); this discovery provided the identity of the CD28-stimulators for the two-signal hypothesis. Shortly thereafter came the discovery of the CD28-like CTLA-4, which was initially, and mistakenly, thought to be another costimulatory receptor, but was soon shown to be an inhibitory receptor by demonstrating its effect as a Fab in mixed lymphocyte reaction (MLR) assays. The inhibitory nature of CTLA-4 was proven following the observation of profound and fatal lymphoproliferation in CTLA-4 knockout mice. After cancer immunotherapy received a critical revitalization following the observation that RAG-2-deficient mice do indeed have more spontaneous tumors, the in vivo pre-clinical and then clinical results showing that anti-CTLA-4 therapy led to very durable disease responses in a subset of patients birthed the modern era of cancer immunotherapy. Searching for new B7 homologues led to the identification of PD-L1 and PD-L2, which were again dogged by the distinction between immune stimulators or inhibitors, and were again proven to be inhibitors by demonstration of Fab activity in MLR assays. Anti-PD-1 and anti-PD-L1 moved rapidly to approval and are now the dominant immunotherapy, with many clinical studies testing combinations with other immunotherapies, with targeted therapies, and with conventional anti-cancer therapies. Although many critical questions remain (why do only some patients respond; defining T cell exhaustion; identifying optimal combination therapies; resistance; biomarkers), Freeman closed with his statement of enthusiasm, confidence, and inspiration for the conference: “With this success, human creativity has been unleashed, and we’re looking to do better”.
Building from the known unique epigenetic state of exhausted T cells in chronic viral infection, Debattama Sen from the Dana-Farber Cancer Institute (Arlene Sharpe and Nicholas Haining labs) began by describing how using ATACseq to assess chromatin accessibility demonstrated the commonality of the core exhausted gene program across both chronic infection and cancer, and how the progenitor exhausted signature and terminally exhausted signature in chronic infection mirrored those in cancer, all driven by the open chromatin state of Tox. Within the chromatin accessibility data, an open chromatin region within the PD-1 regulatory region was present across progenitor and terminally exhausted cells in both tumors and chronic infection, but absent in acute infection. Given the complex and balanced role of PD-1 in both homeostasis and exhaustion, Sen created a germline deletion in this regulatory “enhancer” region to see if the beneficial impact of PD-1 in homeostasis could be preserved while eliminating aspects of exhaustion. Surprisingly, LCMV-specific T cells with the enhancer deletion outcompeted wild-type cells following infection with chronic LCMV without any deficit in functionality, and were more functional (IFNγ and granzyme production) than similar cells from a PD-1 knockout mouse. Compared to wild-type mice, enhancer-deleted mice demonstrated better control of a B16-OVA tumor and enhanced CD4+ and CD8+ (including antigen-specific) T cell subsets. T cells of enhancer-deleted mice showed decreased, but not full loss of PD-1 surface expression, suggesting that reduced, but not absent PD-1 signaling may improve T cell function.
Andrea Schietinger from the Memorial Sloan Kettering Cancer Center probed CD8+ T cell dysfunction in solid tumors by investigating the point at which the T cell exhaustion program is initiated and how the T cell “sees” its corresponding antigen. To study this, Schietinger and her team generated a mouse model for inducible autochthonous liver cancer expressing the SV40 large T antigen to study tumor antigen-specific T cells in the early tumor microenvironment. Using this model, they found that tumor-specific T cells became dysfunctional and began upregulating PD-1 and Lag3 early during tumorigenesis. Interestingly, dysfunction remained relatively consistent between cells taken at day 8 to 10 (early) and cells taken after 30 days or more (late). Early-stage T cells, however, could be functionally reprogrammed into effector cells, and possibly even memory cells, while late-stage T cells were locked into their dysfunctional state. To better understand these functional T cell states, Schietinger observed that the chromatin landscape varied between these two stages (transitioning at around day 10) and that this major remodel is likely irreversible. Next, the researchers investigated which transcription factors govern this fate commitment and found that Tox was associated with loss of TCF1 and with upregulation of coinhibitory molecules on T cells. Overexpression of Tox was sufficient to induce T cell dysfunction in T cells, confirming its involvement. Naturally, the researchers tried knocking out Tox in an effort to improve T cell function, but while Tox-deficient T cells did not upregulate exhaustion markers, their function was still poor, and they did not persist, suggesting that Tox-induced exhaustion programs are actually essential for maintaining viable T cells within tumors. The researchers then uncovered that chronic TCR stimulation induces NFAT signaling, which in turn induces TOX and leads to upregulation of PD-1, Lag3, and CD39, protecting cells from overstimulation and activation-induced cell death. Tox knockout thus represents an interesting scenario in which phenotype is uncoupled from effector function. Schietinger and her team also investigated how TCR affinity affects T cell differentiation and found that within the tumor, T cells containing low-affinity TCRs showed more cytolytic activity than T cells containing medium- and high-affinity TCRs recognizing the same antigen, suggesting that T cell affinity also dictates functional states. This information could be useful in selecting or designing TCRs specific for tumor antigens or neoantigens. Schietinger also presented evidence that knocking out CD8α using CRISPR can lower TCR signal strength and increase antitumor efficacy in vivo.
To uncover mechanisms of acquired resistance to CAR T cell therapy, Marco Ruella from the University of Pennsylvania employed an in vitro genome-wide CRISPR-Cas9 inhibition screen in a B-cell ALL line targeted by CD19 CARs. Long-term (30+ day) co-culture revealed the expected CD19 loss only, and so Ruella turned to short-term (24 hr) co-culture and observed enrichment of genes associated with apoptosis induction by death receptor 5 (TNFRSF10B, FADD, BID, Casp8). Specific knockouts of FADD and BID confirmed this early intrinsic resistance in vitro and in vivo, and the same resistance mechanism was observed in a lymphoma model with an anti-CD22 CAR. Interestingly, BID or FADD knockout led to T cell dysfunction (reduced proliferation, perforin/granzyme production, and cytokine production) in CAR T cells exposed to target during a 15 day culture period, which was confirmed nicely in cell mixing experiments. This led Ruella to hypothesize that following initial elimination of death receptor-sensitive target cells, resistant cells chronically exposed CAR T cells to antigen, leading to T cell dysfunction. Pre-treatment single-cell RNAseq analysis of pediatric B-ALL patient samples from the ELIANA trial revealed a clear distinction between eventual responders and non-responders, with lowered expression of the death receptor signature correlating with poor clinical response. Moreover, RNAseq revealed signals of CAR T cell dysfunction at the time of peak CAR T cell expansion (day 8-10), a signature that was not present in the infusion product. Finally, mimetics of SMAC (a mitochondrial-derived protein which activates apoptosis) such as birinapant pushed the balance toward more apoptosis and sensitized target cells to more effective CAR T cell control.
Aiming to elucidate immune escape mechanisms and to improve the clinical efficacy of T cell therapies, Aude Chapuis from the Fred Hutchinson Cancer Research Center explored optimization of TCR-based treatments in Merkel cell carcinoma (MCC), a cancer that is often driven by the Merkel cell polyomavirus (MCPyV) which encodes an attractive, non-self, viral epitope target. In a clinical trial of 8 patients with MCC who were treated with a triple combination of MCPyV-specific CD8+ T cells, radiation, and anti-PD-L1, 2 patients had a PR and 5 patients had a CR; only 2 patients remain in complete remission. CR was observed in both irradiated and non-irradiated lesions. Mechanistically, triple therapy response involved: MCPyV-specific T cells preferentially localizing to the tumor; epitope spreading, which led to expansion of CD8+ T cell reactivity and induction of de novo MCPyV-specific CD4+ T cell responses; enrichment of activated CD8+ T effectors in the blood; and an increase in intratumoral CD8+ T cells during response. Despite the high percentage of initial responses, most responding patients acquired resistance to the therapy and relapsed. In some patients, the cause of relapse was due to the loss of the targeted HLA. To improve response, Chapuis suggested targeting multiple HLAs to avoid escape, and optimizing the TCR repertoire. To optimize the repertoire, the researchers developed a method of identifying thymus-vetted, high-affinity TCRs from pooled peripheral blood of HLA-matched donors, and sorting them under antigen-specific, tetramer-limiting conditions. Using this method, Chapuis et al. identified a high-affinity TCR targeting an HLA-A*0201-restricted MCPyV peptide, and showed that CD4+ and CD8+ T cells transduced with this TCR efficiently killed MCPyV+ MCC cells in vitro. A clinical trial using autologous T cells engineered to express this TCR has been initiated. Chapuis also suggested enhancing signal 2 by co-expressing an engineered CD8 with intracellular signaling domains (CD28, 4-1BB, etc.) to further improve efficacy and resistance to T cell exhaustion.
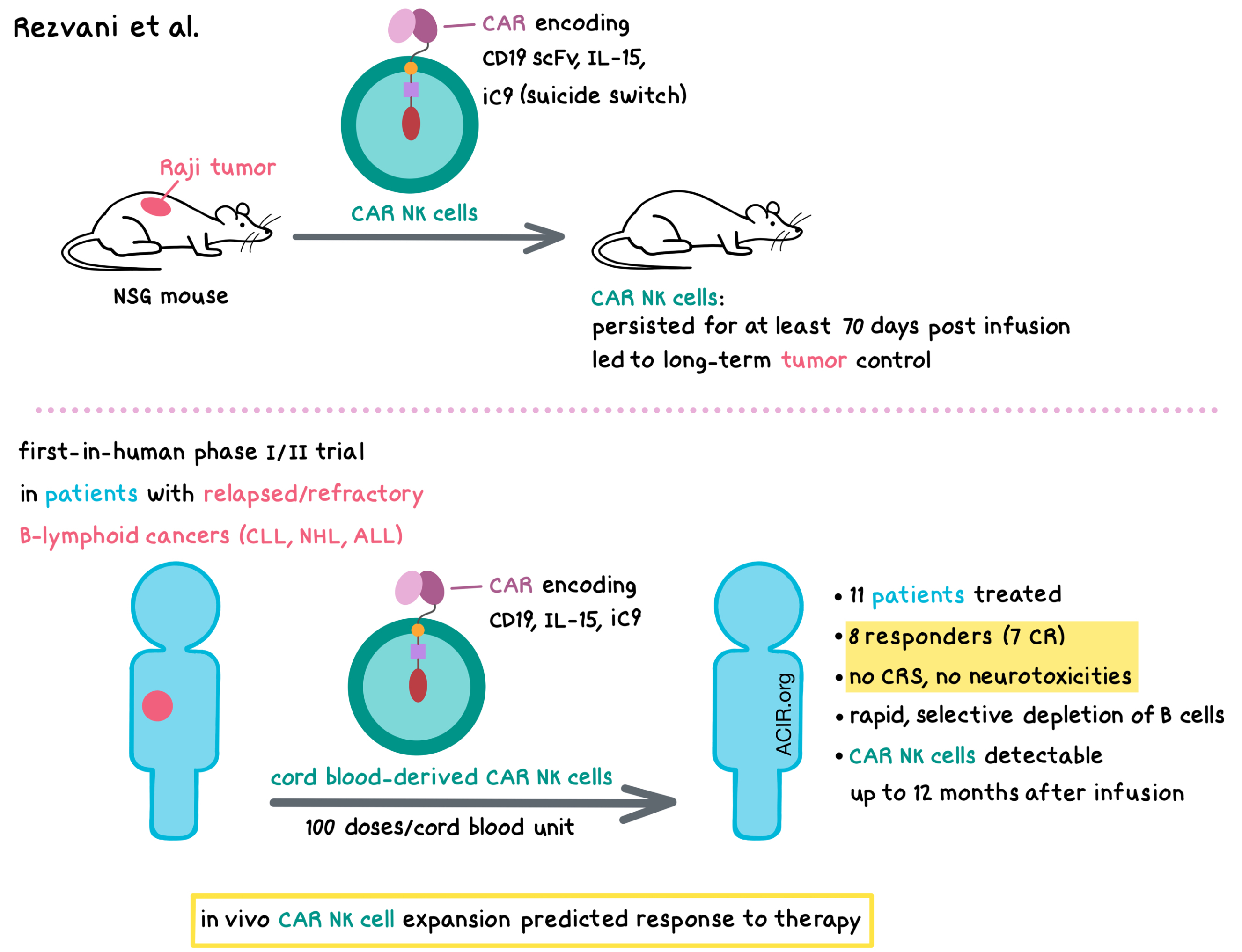
Aware of the challenges that come with CAR T cell treatment (toxicities, target antigen loss, high cost), Katy Rezvani from The University of Texas MD Anderson Cancer Center sought to engineer CAR NK cells for the treatment of hematological and solid cancers. NK cells have advantages over T cells in CAR therapy, including low or no risk of graft-versus-host disease, possibly lower toxicity, target recognition via CAR and NK receptors, the potential to prepare an allogeneic, off-the-shelf product, and possibly lower cost. The limitations of NK cell-based immunotherapy included limited persistence (a couple of weeks) of conventional NK cells and the logistics of sourcing and expanding the NK cells. Rezvani found that off-the-shelf cord blood (CB) is a better source of NK cells than peripheral blood in that CB-derived NK cells have a higher expression of genes involved in the cell cycle, cell division, and DNA replication, and 1 cord unit could provide over 100 doses of NK cells. NK cells transduced with a CD19 CAR construct that also encoded IL-15 and inducible caspase-9-based suicide switch (iC9) persisted for at least 70 days post infusion and led to long-term control of Raji tumors in NSG mice. Rezvani developed a two-week transduction and expansion method for the clinic that generated CB-derived NK cells transduced with a retroviral vector encoding CD19 CAR, IL-15, and iC9, and tested these CAR NK cells in a first-in-human phase I/II clinical trial in patients with relapsed/refractory B-lymphoid cancers (CLL, NHL, ALL). In 11 patients that were enrolled in the dose escalation phase of the trial, no cytokine release syndrome or neurotoxicities were observed. Eight patients responded to treatment, including 7 with a complete response; two patients later relapsed. CAR NK cells rapidly (within 6 days) and selectively depleted B cells and were detectable up to 12 months after infusion. In vivo CAR NK cell expansion predicted response to therapy. An additional clinical trial of CD5-targeting CAR NK cells in patients with relapsed/refractory T-ALL/T-lymphoma is planned.
Discussing opportunities to enhance the efficacy of CAR T cell therapy, Joseph Fraietta from the University of Pennsylvania highlighted the possibility of using epigenetic reprogramming to prevent or reverse T cell-intrinsic resistance. Based on (1) observed differences between responders and non-responders in CAR T cell differentiation, CAR expression levels at peak differentiation, and persistence, and on (2) evidence from a patient in which disruption of TET2 restrained CD8+ T cell differentiation and induced a complete response, Fraietta and his team hypothesized that CAR T cell therapy could be improved by broadly or specifically targeting appropriate epigenetic pathways. To test this, the team used the BET bromodomain protein inhibitor JQ1 to target BRD4 in CAR T cells, which can drive chromatin opening or closing, thereby activating or repressing gene expression. When used in the production of the CAR T cell infusion product, JQ1 increased CAR transgene expression. When used on CAR T cells isolated from non-responding CLL patients treated with CTL019, JQ1 reversed PD-1/PD-L1-mediated immunosuppression, reversed metabolic suppression, and restored cytolytic functions. Transcriptional profiles of these cells revealed upregulation of antitumor response pathways and an increase in genes associated with naive T cell programs. Incorporating JQ1 into the CAR T cell manufacturing process maintained the functional properties of CD8+ T cells, enhanced T cell persistence in vivo, and increased the antitumor efficacy of adoptive cell transfer in mice. JQ1 also enhanced the quality of CAR T cells generated from dysfunctional autologous T cells, which might be useful in expanding the population for whom a high-quality CAR T cell product can be successfully manufactured.
Steven Rosenberg from the National Cancer Institute discussed developing treatment options for metastatic, solid epithelial cancers, which result in over 80% of cancer deaths. He pointed out the advantages and disadvantages of adoptive cell transfer, including CAR T cell therapy, unselected TIL therapy, and selected TIL therapy. Melanoma is currently the only tumor type known to respond completely to unselected TILs, so Rosenberg described the process of selecting neoantigen-reactive T cells using tandem minigenes and autologous antigen presenting cells, which conveniently account for all candidate peptides and MHC loci. Immunogenic neoepitopes have been found to be present in a high portion of patients with many different types of cancer. Extensive data in melanoma and gastrointestinal tumors suggest that about 1.5% of non-synonymous mutations are immunogenic, leading to the generation of both CD8+ and CD4+ T cells. Across all solid cancers tested most neoantigens were entirely unique, with only a few shared in driver mutations. Rosenberg presented data from individual case studies in which selected TIL therapy induced dramatic antitumor results against different cancer types. It may be possible to increase the portion of patients responding to selected TIL therapy by further sorting for markers like PD-1 or 4-1BB, or by transducing mutation-reactive TCRs into naïve or central memory cells. TIL therapies may also be more useful in combination with vaccinations or checkpoint blockades. Generating a library of TCRs against shared antigens, like those arising from driver mutations or “hotspot” mutations, may also help patients by allowing for off-the-shelf products that could be produced faster and at a lower cost than personalized products. Investigating why some TCRs persist longer than others will be useful in understanding and enhancing adoptive cell transfer.
Myeloid Compartment in the TME
Miriam Merad from the Icahn School of Medicine at Mount Sinai called attention to the importance of myeloid cells, as well as to the importance of large-scale research to understand this compartment before jumping into modulating it. Macrophages and dendritic cells populate all tissues, surveying the body, responding to damage or abnormalities, and shaping adaptive immune responses. Dendritic cells and macrophages in tissue may be derived from one of two lineages – from the common myeloid precursors in the marrow, generating long-term resident populations in tissue, or from bone marrow-derived monocytes in the blood. The main theory regarding the difference between these lineages is that tissue-resident macrophages are the first to detect injury, while monocyte-derived macrophages are called in after the fact. To understand the different contributions of these macrophages in tumors, Merad and her team began an extensive immune cell sequencing and mapping effort. Based on data from over 800,000 immune cells, Merad and her colleagues observed that tumor immune responses were strikingly consistent across different patients and different cancers. A fate-mapping murine model system revealed that when tumor lesions were induced, tissue-derived and monocyte-derived macrophages remained transcriptionally distinct, and that blood-derived monocytes could not differentiate into tissue-resident cells. Further, monocyte-derived macrophages dominated the tumor microenvironment while the tissue-resident macrophages tended to accumulate more in healthy tissue. The accumulation of monocyte-derived macrophages was further associated with the accumulation of dysfunctional T cells and, to a lesser extent, with the accumulation of Tregs. In order to target monocyte-derived macrophages without affecting tissue-resident macrophages, the researchers identified high Trem2 expression in the former, but not the latter. Trem2 deletion reduced lung cancer growth and enhanced antitumor immunity, which suggests that strategies to reduce TREM2 in humans may be fruitful. Merad also discussed how CCR2 blockade may prevent macrophage infiltration, and how intermittent fasting regimens may mediate the metabolic activity of monocytes.
Having observed that immune checkpoint blockade is preferentially effective in T cell-inflamed tumors, Stefani Spranger from the MIT Koch Institute for Integrated Cancer Research explored the factors that drive intratumoral T cell infiltration and found that tumors with low T cell infiltration upregulated the WNT/β-catenin pathway and had reduced numbers of Batf3+CD103+ cross-presenting cDC1s. CD103+ cDC1s are the major source of CXCR3 chemokine ligands, and they are required for priming and recruiting T cells into the tumor. However, examination of the intratumoral DC compartments in spontaneously regressing and progressing tumors surprisingly revealed that tumor regression in mice was independent of cDC1s. Knocking out Batf3 in mice showed that T cell induction in regressing tumors was independent of Batf3+ DCs, but dependent on some other CD11c+ DC population. Moreover, the antitumor response was independent of the Sting pathway, but was dependent on type I IFN. Single-cell RNAseq of immune cells from regressing tumors revealed a DC subset that was strongly driven by type I IFN signature and was found in regressing tumors only – Spranger termed this subset interferon-stimulated gene (ISG)-DCs. ISG-DCs were independent of Batf3, stimulated T cells via an unconventional stimulatory mechanism, and were sufficient to drive a systemic antitumor response. In TCGA data, the ISG-DC signature stratified patients’ overall survival in four different cancer types. Thus, Spranger proposed a new working model of T cell activation that includes an alternative danger signal in addition to Sting, engages more than one type of DCs, and leads to non-canonical T cell induction that improves antitumor responses.
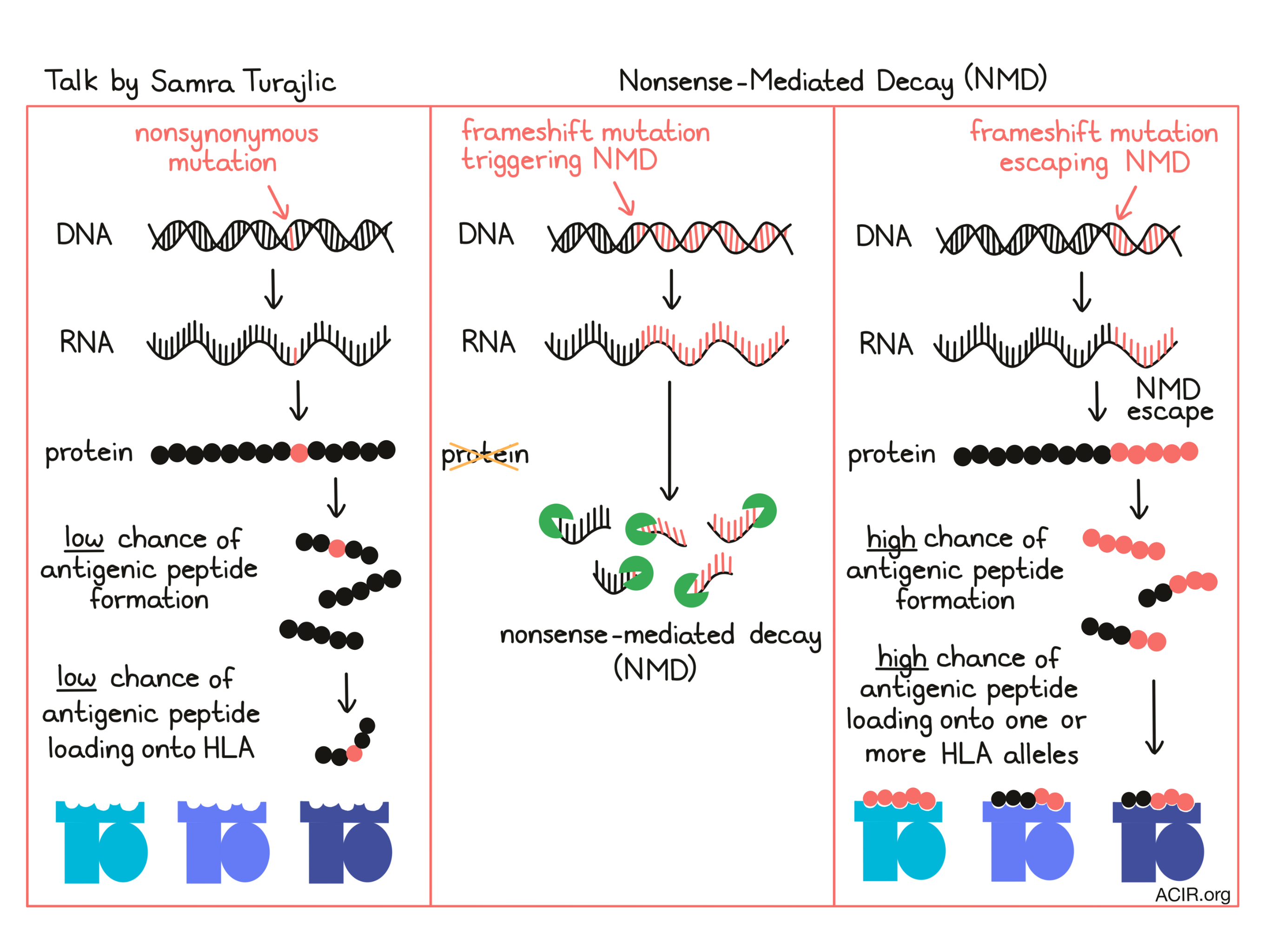
Samra Turajlic from The Francis Crick Institute discussed the role of immunogenic neoantigens in antitumor immunity, as well as the challenges in identifying and targeting them successfully. She noted that while frameshift mutations (indels) make up only about 5% of total mutations, they yield more mutated peptides than non-synonymous mutations, and produce peptides that are highly antigenic. Frameshift indels associate with response to checkpoint blockade in melanoma and, according to some studies, may act as a biomarker. Nonsense-mediated decay (NMD) prevents the accumulation of truncated proteins, but some frameshift mutations, particularly those that occur near the last exon junction, may escape from this regulatory mechanism. Turajlic and her team developed an algorithm to quantify NMD and predict whether out-of-frame indel mutations would escape, resulting in accumulation of NMD-escape neoantigens. Analysis of clinical results showed that having one or more NMD-escape mutation predicted response to checkpoint blockade and adoptive cell therapy. It also correctly predicted patients as responders who would otherwise be classified as non-responders based on tumor mutation burden. Retrospective analysis of human T cell reactivity showed the enhanced immunogenicity of NMD-escape neoantigens, especially those with long (“super”) neo-open reading frames – SNORFs. Given that loss of certain HLA alleles can drive immune evasion, Turajlic and colleagues hypothesized that with a greater number of novel peptides, NMD-escape neoantigens would be able to bind to more HLA alleles, and indeed, a high portion of these frameshift mutations were predicted to bind multiple alleles, and about 10% were predicted to bind to all 6 alleles. Cells with NMD-escape mutations were also found to be under selective pressure and associated with newly primed T cells in NSCLC. Further, NMD-deficient tumors had higher immune infiltrates. Based on this evidence, Turajlic discussed the possibility of pharmacologically targeting NMD escape in cancer – a strategy that is already being actively investigated in germline diseases.
To study the impact of epitope dominance on T cell phenotype, Megan Burger from the MIT Koch Institute for Integrated Cancer Research (Tyler Jacks lab) used the conditional KP lung cancer mouse model, in which tumors express the strong CD8+ epitope SIINFEKL, the weaker CD8+ epitope SIYRYYGL, and a CD4+ epitope from ovalbumin once oncogenesis was initiated. At early time points (8 weeks), intratumoral T cells were increased and tumors were partially controlled, but then rebounded. At 5 weeks, SIINFEKL-specific T cell responses were dominant, but the SIINFEKL-specific T cells were lost over time while the induced SIYRYYGL-specific T cells persisted, eventually reaching comparable levels to the SIINFEKL-specific T cells, but being unable to control tumor. Phenotypically, the SIINFEKL-specific T cells had an exhausted signature, while SIYRYYGL-specific T cells displayed a TCF-1+ “progenitor” signature (reminiscent of the cells shown by Siddiqi et al., 2019 to be responsive to checkpoint blockade). Nonetheless, both cell types responded equivalently to anti-CTLA-4 and anti-PD-1 combination therapy. At early time points, SIYRYYGL-specific T cells showed high expression of CCR6, a chemokine associated with Th17 cells, and experiments with variants of SIINFEKL-specific T cells showed that this CCR6+ phenotype was associated with weaker TCR signaling. Interestingly, vaccination with the SIYRYYGL epitope alone eliminated the TCF-1+CCR6+ population and SIYRYYGL-specific T cells expanded as well as SIINFEKL-specific T cells, suggesting that epitope dominance impacts both T cell phenotype and number.
With the aim to make tumors more immunogenic in situ, Irving Allen from Virginia Tech described studies with irreversible electroporation (IRE) and high-frequency IRE (H-FIRE) – a non-thermal cell ablation approach that creates permanent holes in cell membranes, eventually leading to immunogenic tumor cell death. Although evaluated clinically (in companion dogs and humans) as a tumor ablation strategy, Allen turned to the mouse 4T1 model to study the immune effects of H-FIRE. Treatment of 4T1 tumors with H-FIRE led to a rapid intratumoral decrease in MDSCs, an increase in M1-polarized macrophages, an increase in DAMPs (ATP, ROS, HBG1), and an increase of CD4+ and CD8+ T cells and Th1/Th17 polarizing cytokines. H-FIRE reversed tumor growth beginning ~5 days after treatment and reduced the number of metastatic lesions, consistent with a systemic effect and with elimination of distant metastatic lesions in treated companion dogs. Moreover, treatment of tumor cells in vitro and injection of the resulting cell lysate slowed primary tumor growth and reduced the number of circulating metastatic cells compared to a similar lysate of untreated cells. A key optimization going forward is balancing tissue destruction and immune stimulation.
Joseph Antonios from the Yale School of Medicine presented results from a small (23 patients) phase II trial in glioblastoma using a dendritic cell vaccine pulsed with autologous tumor lysate; patients were randomized to additionally receive resiquimod (TLR7 agonist), poly-ICLC (LTR3 agonist), or no adjuvant. Median survival in the group receiving poly-ICLC was significantly extended (54 months median) compared to resiquimod (15 months) or placebo (11 months). Single-cell RNAseq of peripheral blood samples revealed a significant increase in multiple myeloid populations, as well as a strongly upregulated IFNγ signature only in the patients receiving poly-ICLC. The level of circulating myeloid cells following treatment correlated with survival.
Given that an association between cancer and autoimmune rheumatic diseases has been observed for many years, Antony Rosen from Johns Hopkins University explored the mechanisms behind this relationship and proposed that autoimmune rheumatic diseases may be an example of natural cancer immunoediting. Drawing analogies to biological forces driving Darwinian evolution, Rosen looked to temporal clustering of these diseases and longitudinal trajectory data to seek correlates. Although the clinical clustering of cancer and autoimmune disease is unusual, there is a subset of patients who develop both conditions concomitantly (within 3 years of each other). Furthermore, there have been case reports of patients having their scleroderma improve dramatically with cancer therapy. Rosen found that a close temporal relationship between cancer onset and scleroderma exists particularly in patients with RNA polymerase III (POL3) antibodies, which are autoantibodies found in some cases of scleroderma. Mechanistically, these patients have somatic mutations and loss of heterozygosity at the POLR3A locus, and they bear T cells that recognize both the mutated and the wild-type antigens. Thus, the mutated autoantigen induces both an antitumor response and an immune response to the wild-type form of the autoantigen, leading to major damage of normal tissue. Interestingly, while anti-POL3 autoantibodies were associated with an increased incidence of cancer, other autoantibodies (such as those targeting POLR1 [in the presence of POL3 autoantibodies] or centromere components) were associated with a decreased incidence of cancer. Rosen suggested that rheumatic diseases may provide important insights into natural immunoediting mechanisms that play a role in the treatment of cancer and in side effects of cancer immunotherapies.
Ryan Corcoran from the Massachusetts General Hospital Cancer Center asked whether common oncogenic drivers affect tumor immunogenicity, and found that activation of the RAS-MAPK pathway in tumors leads to sustained proliferative signaling, resistance to cell death, and evasion of immune destruction, making this pathway an attractive immunotherapeutic target. Inhibiting the MAPK pathway using a BRAF inhibitor enhanced the immunogenicity of BRAFV600E melanoma and, in combination with anti-PD-1, prolonged survival in mice. Several clinical trials have demonstrated that targeting BRAFV600E melanoma with a triple combination of PD-1, BRAF, and MEK inhibitors increased the rate of response and progression-free survival (PFS) compared with the BRAF+MEK combination. Similarly, in another clinical trial of patients with KRAS-mutant, microsatellite-stable colorectal cancer (CRC), the combination of MEK and PD-L1 inhibitors led to a 20% response rate with durable responses, whereas no patients responded to either monotherapy. Although these results could not be recapitulated in a larger phase III trial of all CRC types, patient outcomes were stratified by the MAPK gene expression signature – patients with increased MAPK expression had a median PFS of 7-8 months, whereas patients with low MAPK expression had a median PFS of 2 months. Corcoran also explored the resistance to BRAF inhibitors in human BRAFV600E CRC, particularly the secondary resistance that limits the durability of response, and hypothesized that feedback reactivation of MAPK signaling due to emergence of RAS mutations limits efficacy and leads to progression. In fact, MAPK inhibition alone enhanced the immune response in patients with BRAFV600E CRC. Mechanistically, MAPK inhibition led to an increase in intratumoral infiltration of T cells, cytotoxic T cells, and phagocytic cell types. This hypothesis was confirmed in mice with mutant BRAF CRC, where the BRAF/MEK/PD-1 inhibition combination led to a reduction in tumor size and an increase in CD3+ T cells, CD8+ T cells, and CD4+ conventional T cells. These preclinical results led to the initiation of several ongoing trials testing the triple combination in patients with BRAFV600E CRC. Thus, the preclinical and early clinical data suggest that targeting the RAS-MAPK pathway may enhance tumor immunogenicity via increased antigen presentation and induction of various immune signaling programs.
Wendy Garrett from the Harvard T.H. Chan School of Public Health focused on the role of fusobacterium in colorectal cancer. While fusobacterium is normal and expected in the oral cavity, Garret and her team investigated why this bug is so often found infiltrating colorectal tumors and other cancers. Using mouse models, Garrett and her team found that fusobacterium potentiates intestinal tumorigenesis by producing CCL2, which attracts immature myeloid cells and skews their differentiation, leading to selective expansion of myeloid-derived immune cells that suppress antitumor T cell responses. Fusobacterium also directly interacted with tumor-infiltrating lymphocytes to inhibit their functionality, and certain clinical isolates of fusobacteria were able to suppress NK cell cytotoxicity. Sequencing two clones that did not suppress NK cell cytotoxicity identified Fap2 as the gene in fusobacteria responsible for disrupting NK cell function, and other NK cell studies showed that Fap2 binds to TIGIT, a known immune inhibitor that is upregulated in human colon cancer. Exploring how fusobacterium make their way into tumors in the first place, Garrett and her team found that Fap2 again plays a role, mediating fusobacterium enrichment by binding to tumoral sugars. Lineage tracing showed that the oral cavity was likely the initial source of fusobacterium, though whether its path to the tumor is through the blood or the gut is up for debate. Unsurprisingly, given their role in tumorigenesis, fusobacterium infiltration in colorectal cancers was associated with poor patient prognosis. In search of strategies to intercept fusobacterium-infiltrated cancers, Garret and her team found that fusobacterium inhibited Ffar2 signaling in dendritic cells, and that Ffar2 agonism decreased the number of colon tumors, as well as IL-27 expression in dendritic cells and exhaustion in CD8+ T cells. Further, aspirin actually inhibited the survival of some fusobacteria, reducing their abundance in tumors. Other opportunities to target fusobacterium or other microbes relevant in cancer include microbiome replacement, vaccines, antibiotics, and predatory bacteria.
by Anna Scherer, Lauren Hitchings, and Ed Fritsch



