With many CAR T cell strategies in development and successful trials piling efficacy data, the need arises to make these strategies more widely available. Cell manufacturing is costly and demands highly skilled personnel. Methods allowing a less personalized, but rather “off-the-shelf” approach in which CAR-T manufactured from healthy donor T cells can be used are warranted. Recognizing that donor allogeneic CAR-T cells will be rejected by both T and NK cells from the host, or “recipient”, Mo et al. identified and tested a strategy to overcome that rejection. Their results were recently reported in Nature Biotechnology.
Armed with the knowledge that 4-1BB (CD137) is transiently upregulated on recently activated host T and NK cells, and that this would occur as these cells respond to an allogeneic cell challenge, the authors designed a 4-1BB-specific chimeric alloimmune defense receptor (ADR). The ADR consists of the 4-1BB ligand connected via a spacer and transmembrane regions to a signaling intracellular CD3ζ chain. When inserted into T cells, this produced essentially a variant of a first-generation CAR that was able to attack newly activated T cells. T cells could successfully be transduced with the ADR without affecting subsequent expansion. In vitro co-culture experiments confirmed that ADR T cells specifically killed cells constitutively expressing 4-1BBs and newly activated T cells, but were unresponsive to negative controls (resting T cells or cells not expressing 4-1BB). The killing by these ADR T cells was mediated by both Fas-dependent and granzyme B and perforin-dependent pathways.
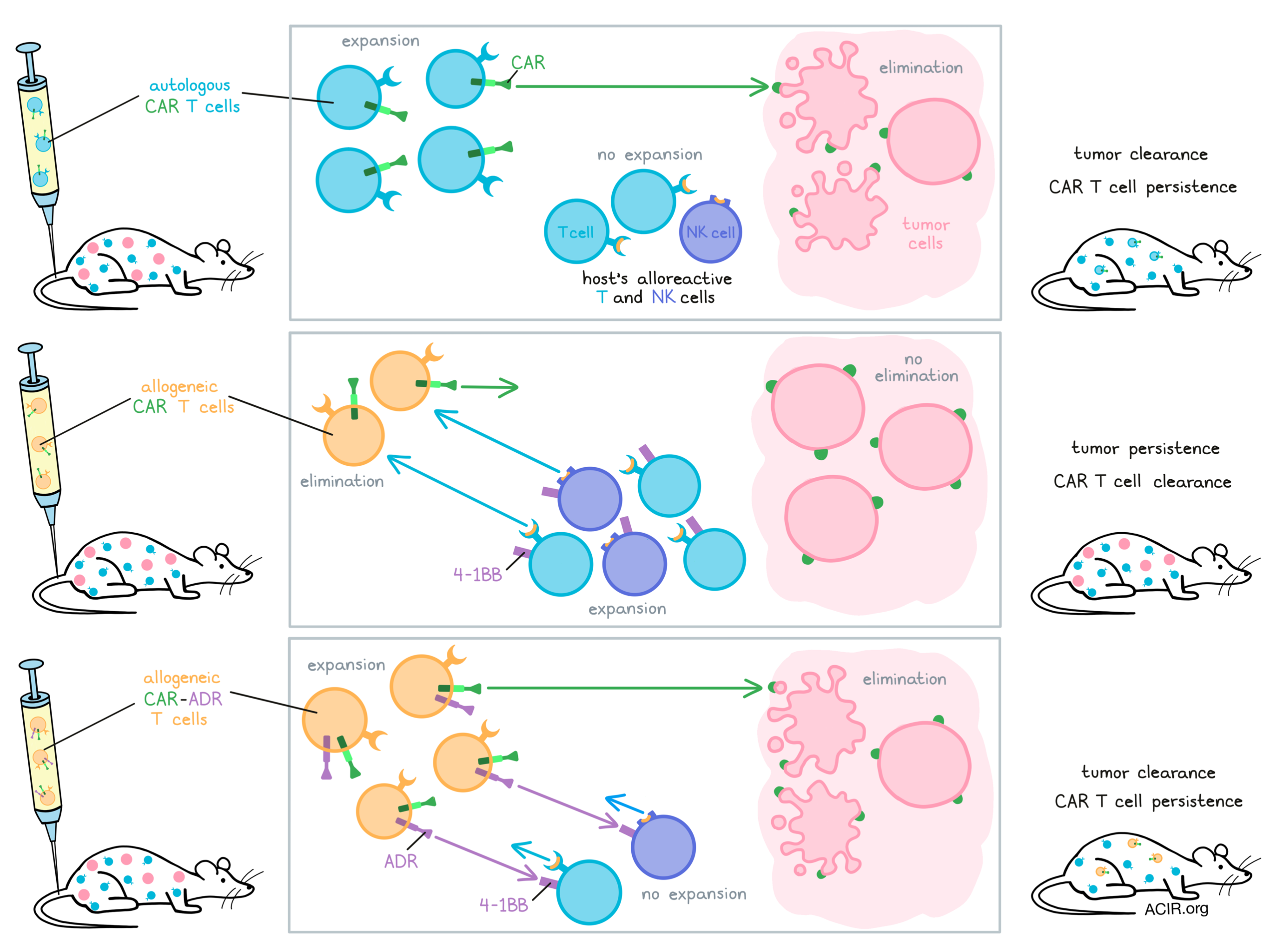
Mo et al. then tested whether ADR T cells could target activated T and NK cells. They developed a mixed lymphocyte reaction model by adding “donor” HLA-A2+ T cells with a Cas9-induced disruption of TCR expression (to reduce graft vs. host alloreactivity) to a 10x excess of PBMCs of HLA-mismatched “recipients”. In this model, control donor cells (no ADR) were eliminated within 9-12 days, while recipient T and NK cells expanded. When ADR-expressing donor cells were used, no elimination took place, donor cells expanded in response to ADR stimulation, and recipient cells were maintained, but did not expand.
To test the targeting of T cell-specific alloimmune rejection, the researchers used NK cell-depleted PBMCs as recipient cells. Again, unmodified T cells were eliminated in 9-12 days, while ADR expression protected the transferred cells from rejection, even when purified, previously primed T cells were added. After confirming that T cells were indeed targeting the donor cells and that ADR prevented such targeting, the researchers moved on to determine whether ADR also protected cells against NK cell-mediated rejection. To do so, they deleted the B2M gene from donor T cells, resulting in MHC-I loss. This actively reduces T cell recognition while promoting NK cell recognition and activation. When control cells were used in this culture system, they were eliminated by PBMCs within six days. Expansion of NK cells indicated that these cells were responsible for the rejection. When the ADR was added to the B2M-edited T cells, the cells were not eliminated, and NK cells did not expand. Similar responses were found when donor cells were cultured with purified NK cells.
To create an in vivo model of alloimmune rejection, sublethally-radiated NSG mice were engrafted with HLA-A2+ recipient T cells (RTCs). Four days later, these mice received control HLA-A2- donor T cells, which were eliminated by day 18. Rechallenging these mice with the same cells also resulted in quick rejection. However, ADR expression allowed donor T cells to survive and expand. An important finding was that not all RTCs were eliminated, and ADR and RTC cells both persisted long-term in these mice, as they did in the in vitro models. To include the effects of NK cells, the researchers substituted the RTCs with whole PBMCs and used mice lacking MHC I and MHC II genes to prevent human PBMCs causing graft-versus-host responses. In this model, unmodified donor T cells were rejected, while ADR T cells not only proliferated and persisted, but also suppressed the expansion of the engrafted PBMCs.
The next step was to combine the ADR with a CAR to establish whether these receptors can function simultaneously and persist in animal models while controlling tumor growth. Using separate ɣ-retroviral vectors encoding each receptor, the researchers induced co-expression of the ADR with a second-generation CD19-CAR on T cells. In vitro co-culture studies confirmed that both CD19+ and alloreactive cells were equally targeted, indicating that CAR-ADR T cells preserve both targeting functions when an ADR and CAR are co-expressed.
To demonstrate tumor control, the authors used NSG mice engrafted with HLA-A2+ RTCs and inoculated them with MHC-I-deleted CD19+ NALM6 acute B-lymphoblastic lymphoma cells. Three days after tumor inoculation, CD19 CAR T cells with or without ADRs were administered. In those mice receiving the CD19-CAR T cells without ADRs, the CAR T cells were rapidly rejected in 19/20 mice, and RTCs expanded in the blood, indicating an active alloreactive response against the CAR T cells. The antitumor effect of the CAR T cells was minimal, and all mice experienced relapses that were eventually fatal. CAR-ADR T cells, on the other hand, expanded in circulation, persisted together with RTCs, and cleared leukemia in 17/19 mice, resulting in long-term survival. Finally, to mimic what would be an anticipated actual off-the-shelf T cell product, CAR-ADR T cells were prepared from TCR-knockout T cells to prevent graft-vs-host activity. Mo et al. confirmed that the use of TCR-knockout CD19-CAR-ADR-T cells also resulted in leukemia eradication in 20/21 mice and prolonged their survival.
CAR T cells may also upregulate 4-1BB on their surface upon antigen recognition and activation, which could potentially lead to self-targeting (fratricide) of the CAR-ADR cells. These in vitro and in vivo data suggest that that does not happen. This may be due to lower availability of detectable 4-1BB on these cells, potentially caused by the masking of 4-1BB through cis binding of the ADR.
Final confirmation of these data was performed in a xenograft solid tumor model of metastatic GD2+ neuroblastoma, in which mice received GD2+ CHLA255 tumor cells and T cells expressing a GD2-CAR with or without ADRs. Again, CAR T cells without the ADR were rejected, resulting in early relapses and severe graft-vs-host disease. However, CAR-ADR-T cells could expand and persist in mice, resulting in sustained eradication of the tumor.
In summary, Mo et al. show that the addition of their ADR to allogeneic T cells shields these cells from both T and NK cell alloreactive elimination. Although further studies are needed to focus on the effects of ADR T cells on other immune populations and on how these constructs affect the risk of cytokine storm or infection, these findings suggest that adding an ADR to a T cell product could pave the way for the use of allogeneic T cells in CAR T cell therapy.
By Maartje Wouters



