
This week’s extensive special feature covers select talks from the SITC - 38th Annual Meeting 2023 in San Diego, CA. We have organized the content by topics below.
Keynote Address
Susan Kaech
Microbiome and metabolism
Susan Bullman
Youngseok Cho
Lydia Lynch
Cell-based immunotherapies
Felix Korell
Elise Wilcox
Leyuan Ma
Kole Roybal
Tumor microenvironment
Rolf Brekken
Bridget P. Keenan
James L. Reading
Humsa Venkatesh
Cancer vaccination
Alex Jaeger
Eduardo Vilar-Sanchez
Hideho Okada
Checkpoint blockade
Lili Yang
Elen Torres
Michelle Tran
David A. Schoenfeld
Cytokines
Karim Chamie
Keynote lecture
Stressing Out T cells in Cancer - Susan Kaech, PhD - Salk Institute
In this Keynote talk, Susan Kaech discussed numerous ways in which stress can shape tumor microenvironments and modulate antitumor immunity. Kaech defined several types of chronic stress that can present in tumors, including mitochondrial stress, oxidative stress, ER stress, nutrient stress, and neuroendocrine stress. Starting with the metabolic stress categories, Kaech described investigations into how alterations in nutrient availability affect T cell function in tumors, and whether T cells adapt to nutrients in a tissue-specific manner. This began with profiling metabolites and nutrients in tumor interstitial fluid, which identified a general increase in many classes of lipids in tumors compared to in circulation. In particular, the researchers noted an increase in arachidonic acid, acyl-carnetines, PUFAs, and oxidized lipids. After discussing several previously identified mechanisms by which lipid classes have been shown to induce T cell suppression, Kaech discussed how T cells adapt to bile acids, which cause ER and mitochondrial stress in T cells in liver cancer. In murine and patient samples, the researchers found that BAAT – an enzyme that conjugates primary bile acids with amino acids – was highly expressed in hepatocellular carcinoma (HCC) samples compared to non-HCC samples. To further interrogate this, the researchers used the AAV-SV40 liver tumor model, which allows for transfer and tracking of antigen-specific CD8+ T cells and comparison with bystander T cells. In this model, antigen-specific T cells showed increased uptake of bile acids compared to bystander T cells, and became exhausted very quickly. Interestingly, different bile acids had different effects on T cells, with only some inducing ROS and killing T cells. This effect could be abrogated by overexpressing a catalase that helps to clear bile acids from the environment. Investigating ways to alleviate bile acid-induced stress, the researchers knocked out numerous targets along the bile acid synthesis pathway, and found that BAAT knockout in particular enhanced T cell infiltration and function, which greatly reduced tumorigenesis. Coming back to human samples, Kaech noted that while most had high BAAT expression, one outlier had low BAAT expression, and this sample was more infiltrated with CD8+ T cells. Across all samples, BAAT expression showed a negative correlation with T cell infiltration, suggesting that altering this profile could be used to enhance T cell infiltration and antitumor immune responses. However, as some secondary bile acids have been known to have positive effects on antitumor immunity, further investigation is warranted. Switching gears, Kaech began to discuss recent research into neuroendocrine stress. In particular, Kaech and colleagues wanted to better understand how stress induced by chronic activation of the sympathetic nervous stress was linked to immunosuppression. Interestingly, when Kaech’s team looked at RNA expression patterns in CD8+ T cells in tumors, they noted expression of the adrenergic receptors Adrb1 and Adrb2, and Adrb1 was particularly upregulated on exhausted CD8+ T cells. This was validated at the protein level, and similar patterns were evident in human data. In a chronic infection model, the researchers found that exhausted CD8+ T cells were localized closer to sympathetic nerves in the spleen, suggesting a relationship between them. Similar patterns were observed in a murine PDAC model, with exhausted PD-1+CD39+ TILs localizing near sympathetic nerves in tumors, and in human NSCLC tumors, where exhausted CD8+ T cells predominated in highly innervated tumor samples. Looking at what happens when Adrb1 is knocked out, the researchers found that the formation of TEX progenitor cells increased, while effector T cells and terminally exhausted T cells decreased. Further, Adrb1 KO T cells showed better viral control, were more responsive to PD-1 checkpoint blockade, and were less localized to peripheral nerves within spleens, suggesting that Adrb1 plays a role in both T cell exhaustion and localization. Finally, the researchers investigated the effects of the FDA-approved beta-blocker, atenolol, which selectively blocks Adrb1. In an immunologically “hot” tumor model, YUMMER, treatment with atenolol alone had little effect, but synergized with anti-PD-1 to enhance T cell cytotoxicity and induce tumor regression. Unfortunately, the same effect was not observed in the immunologically “cold” PDAC model, which remained resistant to anti-PD-1, even with the addition of atenolol. Noting that Adrb2 is expressed on myeloid cells, the researchers treated mice with another FDA-approved beta-blocker, propranolol, which blocks both Adrb1 and Adrb2. In PDAC tumors, this treatment reprogrammed T cell differentiation, increased T cell infiltration and functions, and overcame resistance to anti-PD-1. The combination of propranolol and anti-PD-1 is currently being investigated in clinical trials.
Microbiome and metabolism
Intratumoral Microbes: From Microniches to Single Cells - Susan Bullman, PhD - Fred Hutchinson Cancer Center
Susan Bullman focused on the intratumoral microbiota, and how microbes, immune cells, and malignant cells all interact. Evaluating this, Bullman and team evaluated the spatial distribution of microbes within tumors. In an analysis of both colorectal cancer (CRC) and oral squamous cell carcinoma (OSCC) tumors, Bullman and team found substantial regional variation in the presence of microbiomes within patient tumor specimens. They then adapted a spatial transcriptomics approach (Visium) to identify and map intratumoral microbes, which allowed them to detect the spatial distribution of specific organisms. In some areas of tumors, there were more bacterial cells than tumor cells, while in other areas, microbes were entirely absent. Evaluating the functional differences between these colonized versus uncolonized regions, the researchers found that areas that were heavily colonized were more immunosuppressive, with increased infiltration of myeloid cells, increased expression of checkpoint molecules, exclusion of T cells, upregulation of MAP kinases, and reduced proliferation in these microniches. Investigating whether the bacteria were causing the immunosuppression or simply colonizing a pre-existing niche, the researchers used cancer spheroids to show that in the presence of bacteria, neutrophils were trapped in clusters and upregulated p-ERK and p-p38 in a dose-dependent manner. Further, in the absence of bacteria, the spheroids grew in a single mass, but in the presence of bacteria, cancer cells readily broke off from the spheroid, carrying bacteria with them, suggesting that the presence of bacteria may play a role in increasing cancer metastasis. This was further supported by gene expression analysis, which showed that bacteria-colonized spheroids upregulated pathways associated with epithelial cell migration and metastasis. Interestingly, these cancer cells also had reduced levels of Ki67, in line with a “go or grow” hypothesis. Further, evaluating the bacteria that were carried along with migrating cells, the researchers showed that they adhered to and invaded these cells, where they altered host transcriptional programs at the single-cell level. In particular, invading microbes (dominated by fusobacterium and treponema) largely infiltrated a subset of macrophages and cancer epithelial cells. When the researchers specifically evaluated the effects of fusobacteria on macrophages, they observed upregulation of JAK/STAT and IFN signaling, yet the bacteria remained active, evading the immune cells they had infected. This mechanism of evasion is something that Bullman and team intend to investigate further.
A new oral microbial metabolite prodrug preserves CD8+ T cell stemness and improves the antitumor efficacy of immune checkpoint blockers - Youngseok Cho, PharmD, PhD – University of Michigan
Youngseok Cho discussed how the gut microbiome influences immune checkpoint blockade by mediating T cell metabolism and antitumor immunity. In previous work, Cho and colleagues developed an oral inulin gel (a dietary fiber) that was retained in the colon, altered the gut microbiome, and amplified the antitumor efficacy of anti-PD-1. Metabolic analysis of fecal samples from these mice identified 255 non-short-chain fatty acid metabolites, and pharmacological analysis of top candidate metabolites identified DHB, a major metabolite of polyphenols found in green tea and other dietary sources, for its ability to promote central memory and stem-like memory phenotypes and survival in T cells. Further, treatment with DHB increased IFNγ+ T cells and enhanced IL-2 secretion (specific to the 3,4 DHB derivitive). Metabolomic analysis also indicated the promotion of a progenitor-like and quiescent signature in memory CD8+ T cells. Having identified a microbial metabolite that alters T cell immunity, Cho and colleagues developed a prodrug lipid nano-emulsion formulation of DHB to be delivered orally. In CT26 tumor-bearing mice, oral gavage of this prodrug synergized with delivery of anti-PD-1 to increase tumor antigen-specific T cells, IFNγ responses, antitumor efficacy, and mouse survival. Further analysis of T cell responses showed that DHB increased antigen-specific T cells displaying a stem-like phenotype in tumors, and enriched for central memory T cells in draining lymph nodes. Increased stem cell memory-like (Sca-1+PD-1-) T cells were also observed. The combination of DHB with anti-PD-1 also led to increased recruitment of CD8+ T cells and proinflammatory macrophages; increased secretion of IFNγ, CXCL-10, and granzyme B; and induced a trend towards a reduction in Tregs. Together these results support the possibility that oral delivery of DHB could be used to reprogram antitumor immune responses and enhance cancer immunotherapies.
Diet, Lipids, and Anti-tumor Immunity - Lydia Lynch, PhD – Brigham and Women's Hospital
Obesity is associated with cancer risk, but what remains unknown is how obesity affects immune surveillance. Lydia Lynch discussed that in people or mice with obesity, fewer NK cells were detected, and NK cells had a downregulated cytotoxic machinery and changes in lipid metabolism genes and lipid uptake. NK cells were found to store the lipids, instead of using them for fatty acid oxidation, resulting in metabolic paralysis. While this process did not affect the NK cells’ ability to recognize and migrate to the tumor, it did reduce their antitumor cytotoxic ability by decreasing their ability to degranulate. In tumor models, NK cells from obese donors did not control tumor growth, in contrast to those from lean donors. In endometrial cancer, a cancer type associated with obesity in 50% of cases, patients with a higher BMI had a lower infiltration of CD8+ T cells, suggesting obesity might be associated with a colder immune environment. Besides the immune cells, tumor cells were also impacted by lipids in the environment, and were stimulated towards proliferation and metastasis. Therefore, Lynch investigated whether it was the fat ingested in the diet or the obesity itself that caused the tumor growth. To study this, mice were fed diets with high amounts of different types of animal or plant fats. All mice developed obesity, but tumor growth was increased in mice fed animal fats as compared to plant fats. These effects were not present in RAGγc-/- mice without B, T, or NK cells, suggesting the immune system was involved. NK and CD8+ T cells had lipid accumulation only after an animal fat diet, but not a plant fat diet. Metabolomics analysis to compare the mice suggested that one of the few differences was an increase in octanoyl-carnitine, which is carnitine attached to stearic acid, in mice fed animal fat. T cells can take up long-chain acylcarnitines, which reduces their ability to produce IFNγ and kill tumor cells. On the other hand, acylcarnitines taken up by tumor cells enable immune escape. To investigate whether the stearic acid component of acylcarnitines was boosting tumor growth, Lynch and team assessed cocoa butter, a non-animal fat source high in stearic acid, but a diet high in cocoa butter did not accelerate tumor growth. However, when high levels of cholesterol were added to the cocoa butter diet, there was increased tumor growth. Therefore, a combination of long-chain fatty acids and cholesterol seems to affect tumor growth. These data suggest that diet may have an impact on cancer growth and the antitumor immune response, which could be important during immunotherapy.
Cell-based immunotherapies
Effects of Bcl-2 family protein overexpression in CAR T cells alone and in combination with BH3 mimetics - Felix Korell, MD - Massachusetts General Hospital
Several resistance mechanisms limit the efficacy of CAR T cell therapy in hematological malignancies. Felix Korell, presenting on behalf of Marcela Maus, focused on the resistance of tumors to apoptosis caused by overexpression of anti-apoptotic proteins. All anti-apoptotic proteins contain a common BH3 domain, which has been the target of a variety of BH3 mimetics as antagonists, including Venetoclax (ABT-199), which is approved for treatment of CLL and AML. To be able to combine tumor-targeting BH3-mimetics with CAR T cell therapy and reduce toxicity of BH3-mimetics towards CAR T cells, Korell and colleagues overexpressed Bcl-2 family proteins in CAR T cells. Overexpression of a mutant Bcl-2 (G101V) that is resistant to ABT-199 or a non-mutated Bcl-xL protected 4-1BB-based CAR T cells from elimination induced by ABT-199, and enhanced CAR T cell-mediated killing and CAR T cell expansion in vitro. In long-term proliferation assays in particular, CAR T cells with Bcl-2 (G101V) or Bcl-xL overexpression were less exhausted. In mantle cell lymphoma (JeKo-1) or ALL (Nalm6) mouse models, Bcl-2 and Bcl-xL CARs displayed increased efficacy and persistence, even in the absence of ABT-199. In combination with ABT-199, Bcl-xL CARs displayed significantly increased antitumor activity and survival in the Nalm6 model. Of note, overexpression of both Bcl-xL and Bcl-2 (G101V) in CAR T cells had no additive effect. Bcl-xL outperformed Bcl-2 (G101V) in all experiments and is the top candidate for further analyses.
Identification of alternative splicing-derived targets for TCR-T cell therapies - Elise Wilcox, PhD - Fred Hutchinson Cancer Center
With the goal of identifying more and better TCR-based therapies, Elise Wilcox described her approach to focus on alternative and non-canonical mis-spliced targets (and hence novel junction peptides) in tumors with mutations created by dysfunctional splicing factors. SRSF2 is a key component of the complex, multi-component spliceosome, and recurrent mutations in SRSF2 are commonly found in a variety of cancers, with particular enrichment in myelodysplastic syndrome and chronic myelomonocytic leukemias. These mutations alter the balance of CCNG/GGNG target sites utilized, and result in hundreds of differentially spliced transcripts, some of which are in key hematopoietic gene regulators driving the pathology of the disease, e.g. nonsense-mediated decay of EZH2. To identify which differentially spliced products are presented, Wilcox turned to functional immunopeptidomics with the ARTEMIS method. Cells expressing either wild-type SRSF2 or the dysfunctional mutant (P95H) were transduced with a tagged single-chain dimeric MHC-I allele (HLA-A02, HLA-A11, or HLA-A24, representing ~71% of the US population). Capturing the tagged single-chain dimer allowed MS identification of processed and presented peptides specific for each HLA allele. Bulk RNAseq analysis using rMATS to look for alternatively spliced transcript junctions and translation to peptides provided a database of potential alternatively spliced peptides. This approach identified hundreds to over a thousand peptides per allele, 10% of which were specific to the mutation, and about 50% of which were mutation-associated. As a proof of principle, a number of peptides were processed through an in vitro stimulation process using healthy donor-derived APCs and T cells, and three 2-week stimulations before identifying T cells that were specifically stimulated by the cognate peptide. Two peptides were positive, both of which were derived from alternative exon junctions. To improve the numbers and characteristics of identified peptides, Wilcox focused on non-canonical splicing events, such as retained introns, mutually exclusive exons, alternative 5’ or 3’ splice sites, or skipped exons. This reduced the numbers of events to more comprehensively manageable tens of peptides per allele. Interestingly, although all such peptides were presented (having been identified by immunopeptidomics), many of the peptides were predicted to be weak binders or non-binders, indicating the limitations of predictive algorithms. Combining datasets allowed the classification of the frequency of normal versus altered (tumor) splicing events for each peptide, and two peptides were identified that were highly differentially produced (one due to a retained intron, and one due to a mutually exclusive exon), which may indicate a better safety profile. This pipeline is currently being applied to SRSF2 (P95H) patient samples to identify and confirm novel neoantigen targets for TCR discovery.
Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity - Leyuan Ma, PhD – University of Pennsylvania
Leyuan Ma discussed a synthetic MHC-independent booster vaccine to improve CAR T cell responses in solid tumors. Given that CAR T cells recognize a ligand directly displayed on the cell surface, instead of an antigen presented by MHC, vaccine development to stimulate CAR T cells needs to ensure that the ligand recognized by the CAR is displayed on the APC cell surface. To do this, Ma and team made use of a lipid polymer (Amph-Ligand). This polymer has three major components: a lipid tail, a PEG linker, and the ligand recognized by the CAR. These polymers were formulated into lipid nanoparticles, which, when encountering albumin after injection into mice, falls apart into a monomer and is carried by albumin into the draining lymph node (dLN). In the dLN, the monomer is captured by APCs and is inserted on the cell surfaces. This allows CAR T cells to recognize it as a surface ligand, and together with a signal from an adjuvant (to stimulate APCs to produce signals 2 and 3), it will provide optimal CAR T cell stimulation. An initial test with subcutaneously injected Amph-FITC showed localized cell surface expression on professional APCs in the interfollicular region of the dLN. To model this in a tumor-specific vaccination setting, a glioblastoma model expressing the tumor-specific epitope pepvIII of EGFRvIII was treated with EGFRvIII-CAR T cells combined with an Amph-pepvIII peptide vaccine. The vaccine enhanced EGFRvIII-CAR T cell expansion, persistence, and functionality in peripheral blood. The vaccine also increased the tumoral infiltration, proliferation, and functionality of CAR T cells, and enhanced gene pathways associated with metabolic programming. The vaccine plus CAR T cell strategy cured 60% of animals, and cured mice rejected a tumor rechallenge, suggesting that memory responses were formed. This rejection occurred not only for antigen-expressing tumor cells, but also antigen-negative tumor cells, suggesting antigen spreading-induced tumor-specific endogenous T cells. Similar experiments in WT and Rag1-/- mice confirmed that endogenous T cells were essential for the antitumor efficacy of this therapy in tumors with antigen heterogeneity. Also essential for the efficacy was IFNγ, as IFNγ produced by CAR T cells stimulated DCs to secrete IL-12, which can act on the CAR T cells to continue supporting their functionality. IFNγ also directly acts on CAR T cells, helping to maintain their functionality.
Towards the Develop of Synthetic Immunity to Cancer - Kole Roybal, PhD - University of California, San Francisco
Kole Roybal and team work at the intersection of disciplines to build new receptors and signaling systems that can ramp up the therapeutic function of immune cells and reduce the toxicities associated with cell therapy, especially in the treatment of solid tumors. Previously, they had created a versatile logic-gated receptor system, synthetic notch (SynNotch) receptors, which upon binding a first ligand, release a transcription factor that can drive the expression of a gene, e.g., a CAR that can sense a secondary antigen at the site of disease, reducing on-target/off-tumor toxicity (see also talk by Hideho Okada). Roybal used ALPPL2 – which is highly tumor-specific, but heterogeneously expressed on a variety of solid tumors, including ovarian cancer and mesothelioma – as a priming antigen to upregulate a CAR targeting more homogeneously expressed tumor-associated antigens that otherwise have on-target/off-tumor potential. This circuit not only regulated CAR expression, but reduced exhaustion and maintained the stemness of the CAR T cells by reducing tonic signaling. A therapeutic based on such a SynNotch circuit is currently being tested in patients with ovarian cancer. Next, Roybal broke down the receptors into their various regulated parts, and made large libraries of these components to build thousands of fully human, clinically optimized receptor circuits, synthetic intramembrane proteolysis receptors (SNIPRs), that varied in sensitivity to antigen or transcriptional regulation (e.g., facilitating IL-2 production for autocrine or paracrine signaling). A SNIPR primed by APPL2, regulating the expression of an anti-HER2 CAR, selectively cleared out dual-antigen tumors expressed on one side of the mouse, but did not affect single-antigen (HER2+) tumors on the contralateral flank. Arsenal Bio (co-founded by Roybal) has recently started a clinical trial investigating an APPL2/SNIPR circuit regulating a mesothelin-specific CAR in patients with ovarian cancer. Beyond sensing cell surface antigens, SNIPRs can also be built to sense soluble cues in disease environments, such as cytokines, other molecules found in the extracellular matrix, or synthetic ligands. Advanced engineering of these designer ligand–receptor pairs enables the instruction of cells in vivo to produce therapeutics. While ligand binding induces two cleavage events in the original SynNotch receptors, SNIPRs sensing soluble ligands do not require extracellular ADAM proteases and only cleave in the lysosome. In vivo, SNIPRs sensing natural levels of TGFβ or VEGF in the tumor bed and inducing the expression of a highly toxic HER2-CAR demonstrated antitumor efficacy and prevention of toxicity. The engineering possibilities and customized control of cell therapeutics with SNIPRs seem endless, and Roybal presented the options of creating on/off-switch SNIPRs, conditionally activated SNIPRs, or SNIPRs regulated by dimerization.
Tumor microenvironment
Immune Regulation by Stromal Cells - Rolf Brekken, PhD - University of Texas Southwestern Medical Center
Rolf Brekken discussed work on pancreatic ductal adenocarcinoma (PDAC), which has a tumor microenvironment (TME) characterized by prevalent stromal collagen that impacts tumor progression. Cancer-associated fibroblasts (CAFs) are major drivers of desmoplasia and immune evasion. Brekken found that blocking murine stromal TGFβR2 in a human PDAC xenograft model reduced metastasis, which was related to IL-6 downregulation. IL-6 blockade could also reduce tumor size and metastases, likely due to IL-6’s inhibitory effect on NK cell activity. CAFs were found to produce IL-6 in a TGFβ-dependent manner, driving STAT3 signaling in tumor cells and immune suppression. Characterization of murine PDAC CAFs using scRNAseq identified three clusters, of which one cluster expanded as cancer progressed. This cluster was characterized by expression of mesothelial and MHC-II genes, representing putative antigen-presenting CAFs (apCAFs). Lineage tracing of mesothelial cells during pancreatic cancer progression suggested that these apCAFs were derived from mesothelial cells. Confirming this, PDAC-conditioned media induced an apCAF phenotype in mesothelial cells in vitro. Further analysis showed that IL-1 and TGFβ were sufficient to induce apCAFs from mesothelial cells. To determine whether the MHC-II on these cells was functional, apCAFs were cultured with OVA and OT-II CD4+ T cells. apCAFs do not express co-stimulatory molecules, and in their presence, there was an induction of CD25, CD69, and Foxp3 on the T cells, suggestive of a Treg phenotype. Therefore, apCAFs may contribute to tumor immune evasion by driving Treg formation in an antigen-dependent manner. Murine tumors had an influx of CD3+ and CD8+ T cells and a reduction in Foxp3+ cells after TGFβR2 blockade. However, TFGβR2 blockade also increased tumor cell proliferation and led to worse survival outcomes. Therefore, targeting TGFβ in the TME is of interest, but is challenging. A potential therapeutic strategy to selectively inhibit stromal TGFβ signaling is to genomically stratify patients and only treat tumors with a mutation in canonical TGFβ signaling (TGFβR2 or SMAD4 loss of function mutation, seen in 10% or 50% of PDAC tumors, respectively). Future research will focus on the best way to inhibit stromal TGFβ to enhance the efficacy of both standard therapies and cancer immunotherapy.
Neoadjuvant CD40 agonism remodels the tumor immune microenvironment in locally advanced esophageal/gastroesophageal junction cancer - Bridget P. Keenan, MD, PhD – University of California, San Francisco
Bridget Keenan performed deep immune profiling to define the mechanism of action of the CD40 agonist sotigalimab by examining the effect of a single dose of sotigalimab during neoadjuvant therapy of esophageal/gastroesophageal junction cancer. In this cancer type, neoadjuvant chemoradiation (CRT) is currently the standard practice, and there is a clinical need for increasing the number of pathological complete responses (pCR), as this is associated with survival. In a Phase II trial, a single dose of sotigalimab alone followed by sotigalimab together with neoadjuvant CRT resulted in 38% pCR in this cancer type, which compares favorably to historic controls. Immune profiling of biopsy and resected tumor samples taken post-sotigalimab, but prior to CRT revealed that sotigalimab induced tumoral immune infiltration after a single dose, particularly of T cells and macrophages. The increase in T cells was mostly seen in patients who achieved pCR. Assessing the phenotype of immune cells after sotigalimab treatment showed increased numbers of DCs and increased expression of CD86 and MHC-II on these DCs, suggesting an activated phenotype. At baseline, there were many Tregs in these samples, but these decreased after treatment, while there was an expansion of memory and activated CD8+ T cells. Paired RNAseq and TCRseq was performed to determine T cell clones present at baseline and/or post-treatment. This revealed an increase in induced and persistent CD8+ T cell clonotypes after treatment. In patients with pCR, tumors had a higher proportion of T cells pre- and post-treatment, suggesting a more favorable TME pre-treatment, while tumors not achieving pCR had a higher proportion of myeloid cells. Therefore, this therapy may convert immune cold tumors to hot, potentially by activating intratumoral antigen presentation and reducing suppressive cells, leading to the induction of novel T cell clonotypes and enhancement of T cell activation.
Early remodeling of CD4 T cell differentiation in lung squamous carcinogenesis - James L. Reading, PhD – Cancer Research UK Lung Cancer Centre of Excellence
Lung squamous cell carcinoma is a common cancer type in smokers. James Reading hypothesized that increased antigen exposure, chronicity, and inflammation give rise to a program of T cell differentiation, resulting in the expansion of targetable dysfunctional CD8+ T cells and Tregs. These effects may be paralleled in the periphery, which can then be used for immune-focused early detection. A bronchoscopy surveillance study was performed on heavy smokers to obtain pre-malignant lesions by laser capture for multi-omics T cell analysis. This revealed an enrichment of highly suppressive BATF+ Tregs in these lesions. These highly suppressive OX40hiGITRhiCD177+ Tregs have previously been associated with immunotherapy resistance. To determine how to target these Tregs, a carcinogen-driven model of pulmonary carcinogenesis was developed, in which BATF+ Tregs expanded during carcinogenesis in a similar fashion to human tumors. Gene set enrichment analysis on the Tregs from human and murine samples, performed to look for potential pharmacological targets, showed upregulation of the PI3K pathway in BATF+ Tregs. Treatment with a PI3Kδ inhibitor in the previously described mouse model resulted in 50% fewer mice developing tumors, and those tumors that did develop were smaller, with fewer Tregs and more CD8+ T cells. To track these cells, a high-dimensional analysis was performed on blood from a cohort of patients undergoing bronchial surveillance who eventually developed pulmonary tumors. A peak of CD39+ effector Tregs in the blood was identified, specifically at the inflection points for clinical risk of high-grade preinvasive neoplasia. These Tregs were clonally related to the BATF+ Tregs in the lesions. There was also a spike in TCR clonality that was unrelated to these Tregs, but might track with effector CD4+ or CD8+ T cells. These data were combined to develop a prototype multi-omics T cell early detection index. In a small cohort, this index could forecast survival, showing patients who are unlikely to develop lung cancer if they have a low level of effector Tregs and low TCR clonality in the blood.
The Neural Regulation of Cancer - Humsa Venkatesh, PhD – Harvard Medical School
Turning to the relatively underexplored area of tumor–neuronal interaction, Humsa Venkatesh described work in central nervous system glioma and secondary brain metastatic models, as well as preliminary efforts in the peripheral nervous system. Primary gliomas have long been known to be surrounded by neurons, even in the tumor core. Using an optogenetic model based on blue light stimulation of channelrhodpsin 2 (ChR2) of neuronal circuits demonstrated that stimulation led to increased tumor cell proliferation, and this was dependent on a known synaptic signaling molecule NLGN3. This signaling was further explored and confirmed using EM, electrophysiology, and Ca++ flux, supporting both paracrine and direct signaling mechanisms. As secondary brain metastases (potentially on the rise as short-term control of primary tumors increases) are a predictor of poor outcomes, Venkatesh focused attention on small cell lung cancer (SCLC), a tumor of neuroendocrine origin that has a high rate of brain metastases development. RNAseq showed upregulation of neuronal gene expression, and, interestingly, microscopy of tumor cells showed neural-like cell morphology. Within tumors, closer distance between tumor and neuronal cells correlated with higher tumor cell proliferation and in vitro co-culture of tumor and CNS-derived neuronal cells similarly enhanced proliferation. Stimulation of neuronal cells in the optogenetic model led to enhanced tumor proliferation in vivo, and electron microscopy revealed the presence of both synaptic (tumor:neuronal cell direct apposition) and peri-synaptic (tumor cells adjacent to the interface of two neurons) interactions. Of note, perisynaptic interactions have been previously observed in a breast cancer metastatic model. Bulk RNA sequencing revealed upregulation of key genes involved in ion channels, and single-cell sequencing demonstrated enrichment of this signature in a very particular subcluster of cocultured SCLC cells. In addition to these glutamatergic responses, GABA directly stimulated membrane depolarization of SCLC cells. This effect was opposite the normal depolarization inhibitory role of GABA, but could be explained by a high intracellular chloride concentration, which reverses the GABA signaling. Insertion of the optogenetic ChR2 gene directly into an SCLC cell line demonstrated that blue light-driven stimulation of membrane depolarization enhanced tumor growth. Asking whether the neuronal signaling potential of tumor cells might impact neuronal activity, especially given the observed perisynaptic cell complexes observed, Venkatesh showed upregulation of an astrocytic gene signature in a subcluster of SCLC cells, and morphological evidence of formation of astrocytic-like clustering around neurons. This cellular interaction induced depolarization in the neuronal cells both in vitro and in vivo. Finally, Venkatesh showed that peripheral nervous system interactions can impact tumor growth. Experimental surgical resection of nerves entering the lung (vagotomy) in a GEMM SCLC model led to short-term (100week) and dramatic long-term (6-month) reduction in lung tumor growth, including reduction of liver metastases, and very significantly impacting survival, highlighting the importance of neural cell:tumor cell interactions as an important axis to be explored.
Cancer vaccination
Evaluating MHC-II antigen presentation in vivo to identify targets for immunotherapy - Alex Jaeger, PhD - Moffitt Cancer Center
While mass spectrometry and immunopeptidomics have made strides in unraveling how and what antigens are presented in tumors, Alex Jaeger and colleagues wanted to know how external stimuli in the tumor microenvironment influence antigen presentation, particularly antigen presentation on MHC-II, which is less studied that MHC-I. To explore this, Jaeger developed genetically engineered mouse models (GEMMs) with cre-inducible tags on intact MHC-II on tumor cells (similar to a previously established model for MHC-I). Purification of tagged MHC-II from tumors induced in the KP/AbStrep model allowed for the isolation of MHC-II specifically from tumor cells, as well as immunopeptidomic analysis of what peptides were being presented. The peptides they were able to identify from this model exhibited length distributions that were consistent with peptides presented on MHC-II, validating their system. Comparison of the source proteins for detected peptides bound to MHC-I or MHC-II showed that they were highly divergent, as few proteins gave rise to peptides that were presented on both MHC-I and MHC-II. Peptides from cytoplasmic proteins were equally represented on MHC-I and MHC-II, while peptides from membrane-bound or secreted proteins were favored (about 2:1) on MHC-II, and nuclear or ER proteins were favored (about 2:1) on MHC-I. Interestingly, when the researchers investigated the source of secreted proteins that were being presented, they found that some LUAD MHC-II peptides were exclusively expressed in other TME cell types, suggesting that tumor cells can present peptides from external sources on MHC-II. Jaeger noted that based on this finding, there could be ways to exploit this mechanism by introducing exogenous antigens to be presented on tumor MHC-II. Looking towards the future, they intend to evaluate whether MHC-II epitopes are dynamic through tumor evolution, whether directly presented MHC-II episodes could influence CD4+ T cells, and which MHC-II epitopes are presented by APCs.
Vaccination to Prevent/Intercept Cancer in High Risk Population - Eduardo Vilar-Sanchez, MD, PhD - The University of Texas MD Anderson Cancer Center
Eduardo Villar-Sanchez discussed the immunogenicity of tumors associated with Lynch syndrome, and vaccine development to prevent tumors in carriers. Lynch syndrome is characterized by mutations in mismatch repair genes (MMR; particularly MutS and MutL) resulting in tumors displaying microsatellite instability (MSI). Although resulting in low levels of tumors of various histologies, colorectal and endometrial cancer present the greatest lifetime risk. Genetic profiling of pre-cancer lesions showed an accumulation of predicted MHC class I and II neoantigens, and immune activation could be observed in these lesions, even with a relatively low level of neoantigen accumulation. Mutation profiling showed that all tumors were hypermutant, and approximately one-third of pre-cancer lesions also had a large number of neoantigens. Therefore, vaccination is a potential strategy for carriers of Lynch syndrome. Nous-209 is an adenoviral/MVA vaccine covering 209 shared neoantigens derived from TCGA data, which is thought to cover the majority of tumors. This vaccine was tested in a therapeutic Phase I study combined with immune checkpoint blockade (anti-PD-1), and preliminary results showed patients were highly reactive to the vaccine, with delays in cancer progression detected. Villar-Sanchez then described a more recent trial in 45 healthy Lynch carriers in a Phase IB/II study for preventative purposes. In this study, participants receive an adenovirus prime and MVA boost 8 weeks apart, and the safety and immunogenicity of the vaccine are assessed using blood draws and tissue obtained from colonoscopy. Preliminary data of the first 10 participants showed that all had immunogenic responses, of which, most were reactive to several of the antigen pools. Another vaccine, TdAV5, is a multi-targeted recombinant adenovirus vaccine targeting three tumor-associated antigens (CEA, MUC1, and Brachyury). A Phase IIB trial with this vaccine in combination with an IL-15 superagonist (N-803) among 140 Lynch carriers with previous premalignant lesions is currently ongoing. The primary endpoint is the cumulative incidence of colorectal neoplasms in two follow-up colonoscopies. If these vaccines prove to be effective and safe, Lynch syndrome might become the first genetic disease for which the phenotype can be prevented.
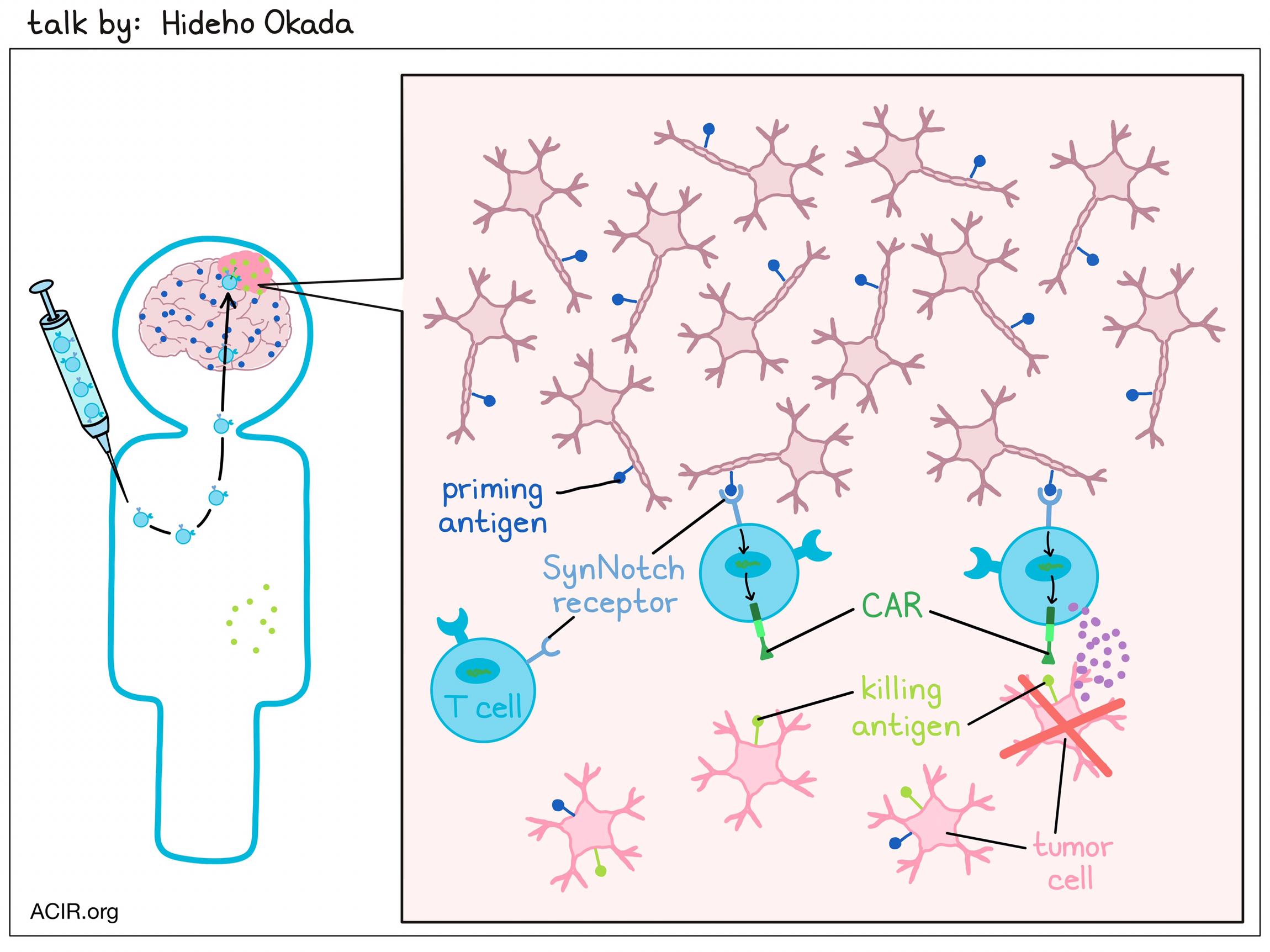
Novel Strategies to Safely Target Brain Tumor Antigens - Hideho Okada, MD, PhD – University of California, San Francisco
To succeed in patients with solid cancer, including glioblastoma (GBM), CAR T cell approaches need to overcome the challenges of on-target/off-tumor toxicities, intratumoral heterogeneity of tumor-expressed antigens, and lack of persistence of T cells due to exhaustion and the effects of tonic signaling. Since there are no perfect antigens for immunotherapy of GBM, Hideho Okada decided to go with two imperfect ones. Okada and team created a "prime and kill" circuit by combining synthetic Notch receptors (SynNotch) that recognize specific priming antigens – which are glioma or brain-specific, but not expressed on all tumor cells – with a CAR that targets glioma-associated antigens, which are more uniformly expressed on tumor cells, but are not tumor-specific. Local recognition of the priming antigen induces expression of the CAR to target the “kill” antigens, leading to local killing of tumor cells, while sparing normal tissue that only expresses the glioma-associated antigen, and not the priming antigen. EGFRvIII-triggered synNotch-CAR T cells targeting EphA2 and IL-13Rɑ2 (E-SYNC) showed tumor region-restricted priming and killing, better efficacy than the individual parental constitutive CAR T cells in heterogeneous GBM patient-derived xenograft models (PDX), and an improved naive/stem-like memory phenotype. Testing different CNS-specific antigens, Okada identified Brevican (BCAN) as the antigen that enabled the most specific and robust priming of the circuit in the CNS. The BCAN synNotch was also combined with a tandem CAR that simultaneously targets EphA2 or IL-13Rɑ2, to generate B-SYNC CAR T cells. A single intravenous infusion of B-SYNC T cells led to sustained complete remission of intracranial patient-derived glioblastoma (GBM6) xenografts, without affecting the growth of BCAN-knockout GBM6 tumors in the periphery, confirming the restricted CAR expression in the CNS. B-SYNC efficiently homed to the GBM site and persisted in the brain. A phase I clinical trial of E-SYNC and B-SYNC T cells is in preparation.
Checkpoint blockade
Beyond the Classical Immune Checkpoint Inhibitors – Targeting the Neuro–Immune interface - Lili Yang, PhD – University of California, Los Angeles
Lili Yang investigated the interface between the nervous system and the immune system in cancer, as both are responsive to external environments and share numerous molecules. To do this, Yang and colleagues extracted TILs from tumors and looked at changes in genes that are known to relate to the nervous system. One gene that stood out was monoamine oxidase A (MAO-A), which was highly upregulated in TILs. MAO-A is an enzyme best known for its role in the brain, where it breaks down neurotransmitters like serotonin, influences mood and behavior, and is the target of a class of antidepressants called MAO inhibitors (MAOIs). Investigating its role in the immune system, Yang found that in mice with MOA-deficient immune systems (global loss of MOA or specific to CD8+ T cells), tumor growth was reduced and antitumor immunity was enhanced. Digging deeper, the researchers found that in wild-type mice, CD8+ T cells upregulated both MAO-A and serotonin upon stimulation. The MAO-A then broke down the serotonin, reducing T cell autocrine serotonin signaling through T cell-expressed 5-HTR (the serotonin receptor), and ultimately creating a negative feedback loop that regulates the CD8+ T cells. To take advantage of this non-traditional checkpoint pathway for cancer immunotherapy, the researchers tested blockade of MAO-A using traditional MAO inhibitors, such as phenelzine, which improved serotonin secretion and induced T cell production of IFNγ in vitro. In vivo, phenelzine alone did not show antitumor efficacy, but synergized with anti-PD-1 in several syngeneic mouse models. Looking for translational relevance, the researchers found that human T cells also upregulate MAO-A upon T cell antigen stimulation, and showed that MOAIs could enhance responses to anti-PD-1 in xenograft mouse models as well. In patient data, low MAO-A was often correlated with poor prognosis. Investigating MAO-A expression in other immune cell types, the researchers found that it was also highly expressed in tumor-associated macrophages, where it guided polarization towards a more M2-like phenotype. However, as macrophages do not make or sense external serotonin, the researchers explored other possible mechanisms, and found MOA-A-mediated macrophage repolarization was related to a buildup of reactive oxygen species. MAOI treatment could revert macrophages back towards a more M1-like phenotype. Together, these results suggest that MAOIs could have multiple beneficial effects in support of cancer immunotherapy, both by releasing a checkpoint on CD8+ T cells and by supporting a more favorable macrophage phenotype. Given that MAOIs are already approved and widely used for other purposes, their implementation in cancer immunotherapy could be a relatively straightforward way to improve outcomes, and certainly warrants further investigation.
SOX2 expression in lung cancer mediates resistance to checkpoint blockade therapy by inducing Treg-dependent CD8+ T cell exclusion - Elen Torres, PhD – Koch Institute for Integrative Cancer Research at MIT
While chemotherapy and immunotherapy can effectively treat a wide range of patients with cancer, many patients still do not respond to currently available therapies. To better understand and overcome this, Elen Torres and colleagues defined types of tumors based on their patterns of T cell infiltration: infiltrated with functional T cells, infiltrated with non-functional T cells, T cell-excluded (T cells only around the tumor edges [peri-tumoral]), and non-infiltrated. Analyzing data for NSCLC from TCGA, the researchers identified T cell-inflamed and non-T-cell-inflamed gene signatures, and saw that Sox2 was negatively correlated with the T cell gene signature. This pattern was also seen in biopsies of lung squamous cell carcinoma and lung adenocarcinoma, which showed high Sox2 expression in tumor regions with low CD8+ T cell infiltration, and vice versa. Evaluating this in an experimental setting, Torres and team used a KP adenocarcinoma to show that when Sox2 was overexpressed, T cells failed to infiltrate tumors or slow tumor growth, suggesting that high Sox2 expression indeed mediates T cell exclusion. Knowing that T cell exclusion is often associated with poor response to immunotherapy, the researchers evaluated anti-PD-1/anti-CTLA-4 and showed that Sox2 overexpression also mediates resistance to therapy. In an effort to improve infiltration of tumor-specific T cells, the researchers utilized a tumor antigen-specific adoptive transfer model to show that while the adoptively transferred T cells are effectively recruited to both WT and Sox2+ tumors, they fail to infiltrate the core of the Sox2+ tumors, remaining at the outer edges. Evaluating how Sox2 might mediate immune exclusion, the researchers noted a high density of Tregs in the peritumoral region of Sox2+ tumors, and gene expression and protein ELISA analyses suggested these Tregs are likely recruited by increased levels of CCL7 and CCL2 via CCR2. Targeting these Tregs using anti-GITR increased CD8+ T cell infiltration into the cores of Sox2+ tumors, and sensitized tumors to checkpoint blockade therapy. Depletion of Tregs also improved the tumor vasculature, which could support improved infiltration of various immune subsets. Together these results suggest that high Sox2 expression mediates increased expression of CCL2 and CCL7, leading to recruitment of Tregs that exclude CD8+ T cells from tumors. Understanding and targeting this mechanism has the potential to improve T cell infiltration and enhance immunotherapies.
Dissecting resistance to immune checkpoint blockade in bladder cancer: single cell analyses link pro-inflammatory macrophages and IL-6 to CD8+ T cell suppression - Michelle Tran - Icahn School of Medicine at Mount Sinai
Immune checkpoint blockade (ICB) is used to treat bladder cancer, but only 20-25% of patients respond to this therapy. To better understand resistance mechanisms, Michelle Tran identified gene signatures in pre-treatment tumor samples related to overall survival. Two main signatures were detected; the signature related to better survival was enriched in adaptive immune genes, while the signature related to worse survival was enriched in innate immune pro-inflammatory genes. To combine these gene signatures, a ratio (adaptive/pro-inflammatory) was created that was called the 2IR score, with high scores being associated with response to ICB and low scores with therapy resistance. To study the underlying mechanisms, bladder tumors were subjected to scRNAseq, revealing monocytes and macrophages as important components of the pro-tumorigenic inflammation signature. Zooming in on the macrophages, Tran found a cluster of macrophages with high expression of SPP1 and a macrophage cluster highly expressing C1QC. The SPP1hi macrophages were enriched in proinflammatory IL-6 and IL-8 signaling pathways and hypoxic signaling pathways, whereas the C1QChi macrophages were enriched in antigen presentation and complement gene pathways. SPP1hi macrophages were associated with ICB resistance, while responders had more C1QChi macrophages. Further characterizing bladder SPP1 tumor macrophages revealed an upregulation of Clec5a and Trem1. IL-1β was detected as the strongest inducer of Clec5a+Trem1+SPP1+ macrophages in in vitro experiments. Higher IL-6 plasma levels also correlated with worse survival in an ICB trial, and this was mostly produced by the SPP1 macrophages. Others have recently shown that IL-6 signaling impairs the differentiation of cytotoxic effector CD8+ T cells, which was confirmed by Tran in this model. Therefore, Tran proposes IL-1β in the bladder microenvironment induces Clec5a+Trem1+SPP1+ macrophages that secrete IL-6, which impacts circulating CD8+ T cells and blocks their differentiation into cytotoxic effector cells.
“Decoy-resistant” IL-18 in combination with CTLA-4 blockade enhances anti-tumor efficacy in preclinical models of renal cell carcinoma - David A. Schoenfeld, MD, PhD – Yale Cancer Center
IL-18 as an immunotherapy often lacks efficacy, which may be due to the induction of IL-18 binding protein (IL-18BP) in the tumor microenvironment. IL-18BP binds and inhibits the activity of IL-18. David Schoenfeld presented data on a “decoy-resistant” IL-18 (DR-18) that can stimulate immune cells, but is resistant to IL-18BP inhibition. In various murine cancer models, DR-18 had activity as a monotherapy, and worked synergistically with PD-1 blockade. In clear cell renal cell carcinoma (ccRCC), there is a high expression of IL-18BP and the IL-18 receptor. Nonresponders to immunotherapy had a significant increase in IL-18BP levels, and these patients had worse survival, suggesting it may be a mechanism of resistance. Therefore, DR-18 might be of interest for the treatment of ccRCC. To test its potential effects, murine RCC models (Renca and RAG) were treated with DR-18 and immune checkpoint blockade (ICB; anti-PD-1, anti-CTLA-4, or the combination). DR-18 or ICB alone had modest effects, and the combination of DR18 and anti-PD-1 did not add benefit. However, the combination of DR-18 and anti-CTLA-4 worked synergistically, and further addition of anti-PD-1 had no or minimal benefit. The DR-18/anti-CTLA-4 combination induced memory responses, and CD8+ T cells, NK cells, and IFNγ were all required for its efficacy, while CD4+ T cells were less important. Cytokine and chemokine profiling showed broad inflammatory responses to DR-18 and anti-CTLA-4, and key early mediators were IFNγ, IP-10 (CXCL10), and MIG (CXCL9). In tumor tissue, there was an increase in granulocytes and CD4+ and CD8+ T cells, and monocytes and macrophages changed to be more proinflammatory. Tumoral CD8+ T cell activity was increased, as was the expression of checkpoints on T cells. There was an enrichment in precursor and terminally exhausted T cells, and a clonal expansion of CD8+ effector T cells. Granulocytes were also enriched after treatment, and neutrophil characterization showed that the antitumor N1 and N2 subtypes increased after combination therapy. Further research in other (ICB-resistant) murine models and testing other therapeutic combinations are underway.
Cytokines
Cytokines in Cancer: Clinical Urological Perspective of N-803 - Karim Chamie, MD - University of California, Los Angeles
Bladder cancer is very common, but thankfully, most are non-muscle-invasive and can be treated with intravesical administration of BCG, which acts as a cancer vaccine and induces both adaptive T cell-mediated responses and innate immune memory through NK cells. While this standard-of-care therapy is effective in a majority of patients, those who are refractory or who relapse following BCG therapy are unlikely to respond to subsequent dosing, and are left with few options. As relapse may be due to HLA downregulation and subsequent T cell evasion, Karim Chamie and others investigated the possibility of using N803, an IL-15 superagonist, to expand NK cells, cytotoxic CD8+ T cells, and CD4+ T cells, and restimulate responses. In mice, the combination of N-803 with BCG treatment induced innate immune memory to overcome T cell evasion, and induced the proliferation of NK cells, T cells, and memory T cells, resulting in long-term memory and durable complete responses. Testing this strategy in the clinic, patients who had not received prior BCG treatment were treated with escalating doses of N-803 in combination with BCG, and in the small cohort, every patient experienced a complete response with no evidence of disease recurrence after 2 years of follow-up. Next, Chamie turned to patients with carcinoma in situ (CIS) or papillary disease, who had previously been unresponsive to BCG therapy, and for whom treatment options are limited and are largely ineffective. Due to the unmet need in these patients, the researchers were able to run a single-arm trial evaluating N-803 + BCG, and found that among 100 patients with non-muscle-invasive bladder cancer with carcinoma in situ, 71% had a complete response, with 62% maintaining that CR at 12 months, and 53.3% maintaining it at 24 months. Overall, the median duration of CR was 26.6 months. Importantly, 90% of patients were able to avoid a cystectomy (89% at 24 months), allowing for a better quality of life. Only 7% of patients had to discontinue treatment due to adverse events. Interestingly, responses appeared to occur irrespective of any evaluated patient factors, including relapsed vs. refractory disease, CIS or papillary disease, prior BCG dosing, or age. In 80 patients with papillary disease, which is surgically removed as the standard of care, 98% of patients treated with N803 + BCG were alive at 12 months, and 93% avoided cystectomy. The disease-free survival rate was 55% at 12 months, 51% at 18 months, and 48% at 24 months. Toxicities associated with treatment were mostly local to the bladder, and were similar to those observed with BCG treatment alone, with only 2% treatment discontinuation due to adverse events. Overall, these clinical data are promising, and could serve to benefit patients with both treatment-naive and relapsed/refractory bladder cancer.
By Lauren Hitchings, Maartje Wouters, Ed Fritsch, and Ute Burkhardt



