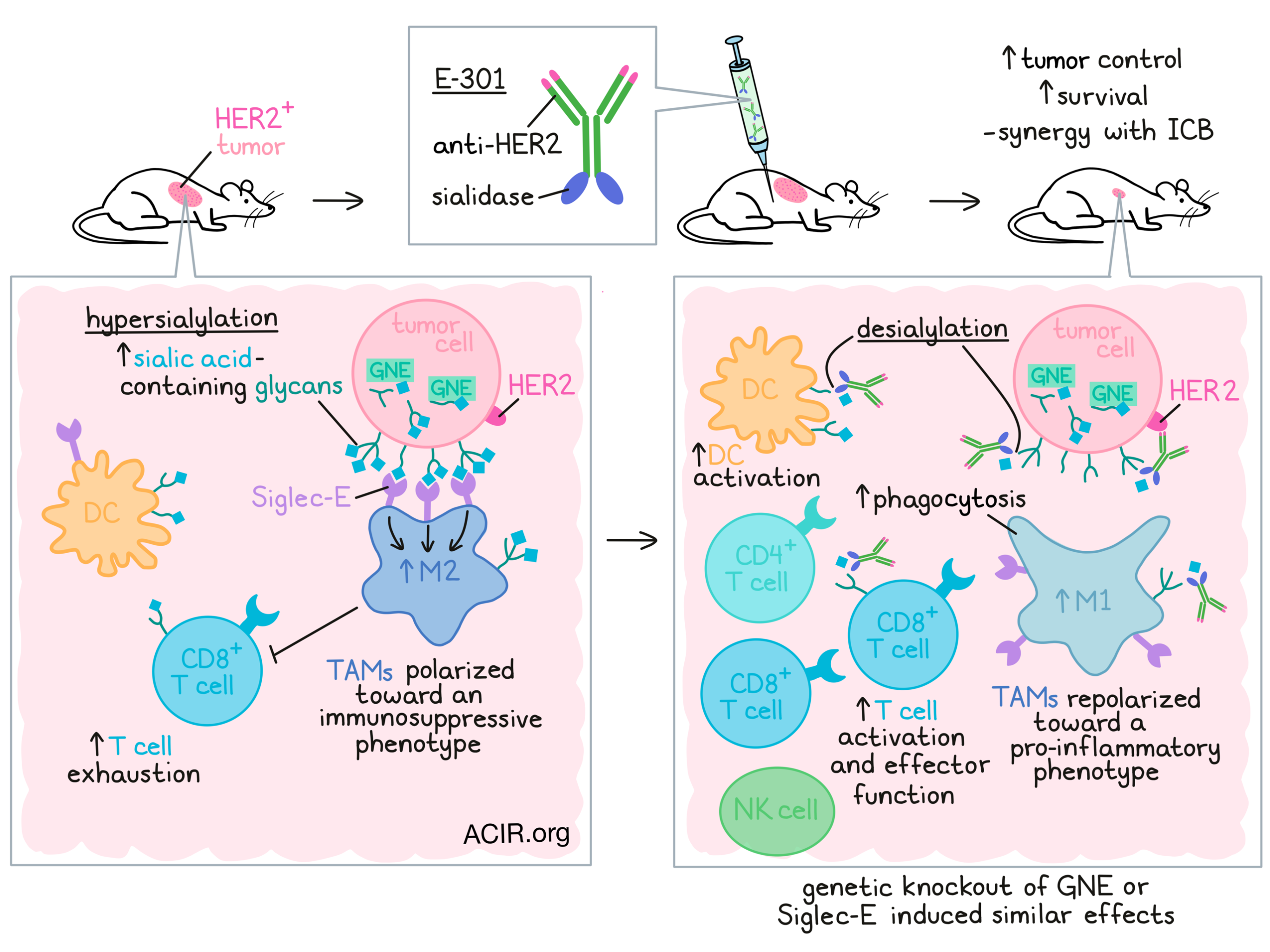
In cancer, upregulation of sialic acid-containing glycans (hypersialylation) is a feature that has been shown to drive disease progression and immune escape through engagement with Siglec receptors and inhibition of tumor-infiltrating immune cells. In previous work, Stanczak et al. showed that genetic desialylation or treatment with sialidase could reduce tumor growth and contribute to antitumor immunity. In work recently published in Science Translational Medicine, Stanczak et al. dug further into the details of this mechanism and explored strategies to exploit it for cancer immunotherapy.
To begin, Stanczak et al. used data from The Cancer Genome Atlas (TCGA) to identify a gene signature of sialoglycan genes that positively correlated with regulation of immune function, reduced cytokine activity, and shorter patient survival in several cancer types, most notably, clear cell kidney cancer (KIRC) and squamous cell lung cancer (LUSC). This gene set was also strongly correlated with the presence of immunosuppressive and pro-tumor immune cells (including Tregs and TAMs), reduced CD4+ T cells, and CD8+ T cell dysfunction. These results were validated in patient samples, where Siglec-9 Fc binding, a measure of surface sialylated glycans, was associated with worse survival (in KIRC), and treatment with sialidase reduced T cell activation (in LUSC).
Moving to mouse models, the researchers utilized MC38 tumors with genetic knockout of GNE, the rate-limiting enzyme for sialic acid biosynthesis. As in previous work, growth of GNE-KO MC38 tumors was delayed and mouse survival was increased compared to wild-type MC38 – an effect that was dependent on CD8+ T cells and could be enhanced with anti-PD-1 and anti-CTLA-4 (ICB). In a repeat experiment in which TILs were isolated on day 7, treatment with ICB alone increased the frequency of IFNγ+ and IFNγ+TNFα+ CD8+ T cells in both WT and GNE-KO tumors, but only induced IFNγ+TNFα+IL-2+ CD8+ T cells in the GNE-KO, suggesting that desialylation could enhance T cell activation in the context of ICB. Similar results were observed using other tumor cell lines and mouse strains.
Next, the researchers evaluated therapeutic desialylation using an an antibody–sialidase fusion product, E-301, comprising a HER2-targeting antibody (trastuzumab) and two sialidase domains fused to the c-terminal Fc region of the antibody. In vitro, E-301 induced dose-dependent desialylation of EMT6-HER2 mammary carcinoma cells. Similarly, in mice bearing established EMT6-HER2 tumors, a single intraperitoneal dose induced desialylation that was most pronounced at 24 hours, and still evident at 72 hours. At this same time point, the tumors also showed a loss of Siglec ligands, further validating the results.
Looking more closely at the effects of desialylation on immune cells, the researchers implanted mice with wild-type and GNE-KO EMT6-HER2 tumors on opposing flanks. The GNE-KO tumors showed reduced sialylation, which could be reduced slightly further with E-301. Treatment with E-301 induced desialylation of Infiltrating immune cells equally on both sides. This effect was observed across a broad array of immune cells, including TAMs, DCs, PMN-MDSCs, Tregs, and CD8+ T cells, but not M-MDSCs, conventional CD4+ T cells, or NK cells.
Stanczak et al. also found that in mice bearing B16D5-HER2 melanoma tumors, treatment with E-301 induced a number of changes in immune infiltrates, with treatment increasing absolute numbers of CD45+ and CD8+ T cells, DC activation, antitumor NK cell subsets, effector CD8+ T cells, and both Th1 and Th2 CD4+ T cells, while reducing naive CD4+ T cells and exhausted CD8+ T cells. Macrophages showed particularly dramatic changes, demonstrating a shift from a more immunosuppressive phenotype to a more pro-inflammatory phenotype, with increased signs of migration, responsiveness to chemokines, and production of effector molecules and pro-inflammatory cytokines. The researchers also noted an increase in M1-like and decrease in M2-like macrophages, altering the M1:M2 ratio. Similar immune changes were observed in the E-301-treated EMT6-HER2 tumor model, and in ICB-treated GNE-KO tumors.
Looking more closely at changes in TAMs, the researchers cocultured tumor-derived TAMs with naive CD8+ T cells and found that T cells cocultured with TAMs from genetically or therapeutically desialylated tumors were more proliferative and activated. Further, when desialylated tumor cells were cocultured with bone marrow-derived macrophages (BMDMs), the BMDMs expressed higher levels of TNFα and an increase in phagocytic activity. These results are in line with sialic acid acting as a “don’t eat me” signal, and suggest that high levels of sialic acid on tumor cells promotes the polarization of TAMs towards an immunosuppressive, pro-tumorigenic phenotype.
Investigating how desialylation can repolarize macrophages, Stanczak et al. showed that among common inhibitory CD33-related Siglecs, Siglec-E was most the most broadly expressed on TAMs, and had the highest expression on anti-tumorigenic TAMs, which were increased in E-301-treated tumors. In Siglec-E-KO mice, GNE-KO tumors grew as well as wild-type, and treatment of various tumors with E-301 was ineffective. Similar results were observed in mice depleted of TAMs via treatment with an anti-CSF1R antibody and in mice with Siglec-E conditionally knocked out on CD11c+ cells – a population that included TAMs, DCs and PMN-MDSCs, but mainly represented TAMs (Siglec-E on DCs showed minimal contributions to the effects). In the Siglec-E conditional knockout model, TAMs showed reduced M2 polarization. In vitro, Siglec-E-KO TAMs did not repolarize or increase activation of cocultured T cells upon treatment with E-301.
Evaluating the potential efficacy of E-301 as an immunotherapy, the researchers showed that in mice bearing orthotopic EMT6-HER2 tumors, 4 doses of E-301 delayed tumor growth and improved survival, with 1 of the 12 mice rejecting the tumor entirely. When this mouse was rechallenged with EMT6 and EMT6-HER2 tumors on opposing flanks, both tumors were rejected, suggesting strong immune memory and induction of response beyond HER2. Similar results were observed in the B16D5-HER2 model, where treatment with E-301 reduced tumor growth dependent on CD8+ T cells and synergized with ICB. Anti-PD-1 and ICB were also more effective in Siglec-E-KO mice, with 14 out of 18 (78%) of Siglec-E-KO mice rejecting tumors, compared to 4 out of 15 (27%) wild-type mice.
Overall, these results suggest that hypersialylation on tumor cells promotes polarization of TAMs towards an immunosuppressive phenotype through interactions with Siglec-E. They also support the sialoglycan-Siglec axis as a target for immunotherapy, as therapeutic desialylation can promote the repolarization of TAMs and support strong antitumor immune responses, particularly in the setting of ICB.
Write-up and image by Lauren Hitchings.
Meet the researcher
This week, first author Michal A. Stanczak and lead author Heinz Läubli answered our questions.

What was the most surprising finding of this study for you?
HL: The most surprising finding of the study was the robustness of the results across multiple experimental models. Removing sialic acids led to improved antitumor immunity, be it by genetic means or using the enzymatic approach.
MAS: For me, the simplicity of the mechanism has been most surprising. We’ve been thinking and speculating about the incredible heterogeneity of glycans and the potential for functional redundancy of Siglec receptors for a long time. And while we can’t exclude that some of that plays a role, the expression of Siglec-E specifically on TAMs turned out to be the single dominant mechanism conferring the immunosuppressive effects of tumor sialylation in our hands.
What is the outlook?
HL and MAS: A whole range of basic and preclinical research supports tumor glycosylation as a potential target for cancer immunotherapy. The next big steps for the field will be the clinical testing of that potential. Early clinical studies delivering human sialidase to patients with cancer are currently ongoing (GLIMMER-01 study). While it remains to be seen if this will ultimately turn out to be the best approach, future investigations will further aim at the exact intracellular events in myeloid cells following therapeutic interference and will compare the efficacies of the different approaches to block sialic acid–Siglec interactions, be it by blocking Siglec receptors or directly targeting sialic acids.
What was the coolest thing you’ve learned (about) recently outside of work?
HL: The coolest thing I have learned recently is that my 9-year-old son can already play better ice hockey than me….
MAS: I recently got into making my own pasta from scratch. Took a bit of experimentation, not unlike the work in the lab, but the results are much tastier.



