Personalized cancer vaccines have demonstrated safety and potency in early clinical trials; however, the magnitudes of T cell responses have been limited, and definitive positive clinical effects have not been demonstrated. These results suggest that improving the magnitude may be beneficial, and that the quality of vaccine-responding T cells must also be considered. As outcomes may depend on the vaccine delivery strategy, Baharom et al. hypothesized that varying the route of vaccine administration could tune the magnitude and phenotype of antigen-specific T cell responses, as recently reported in Nature Immunology.
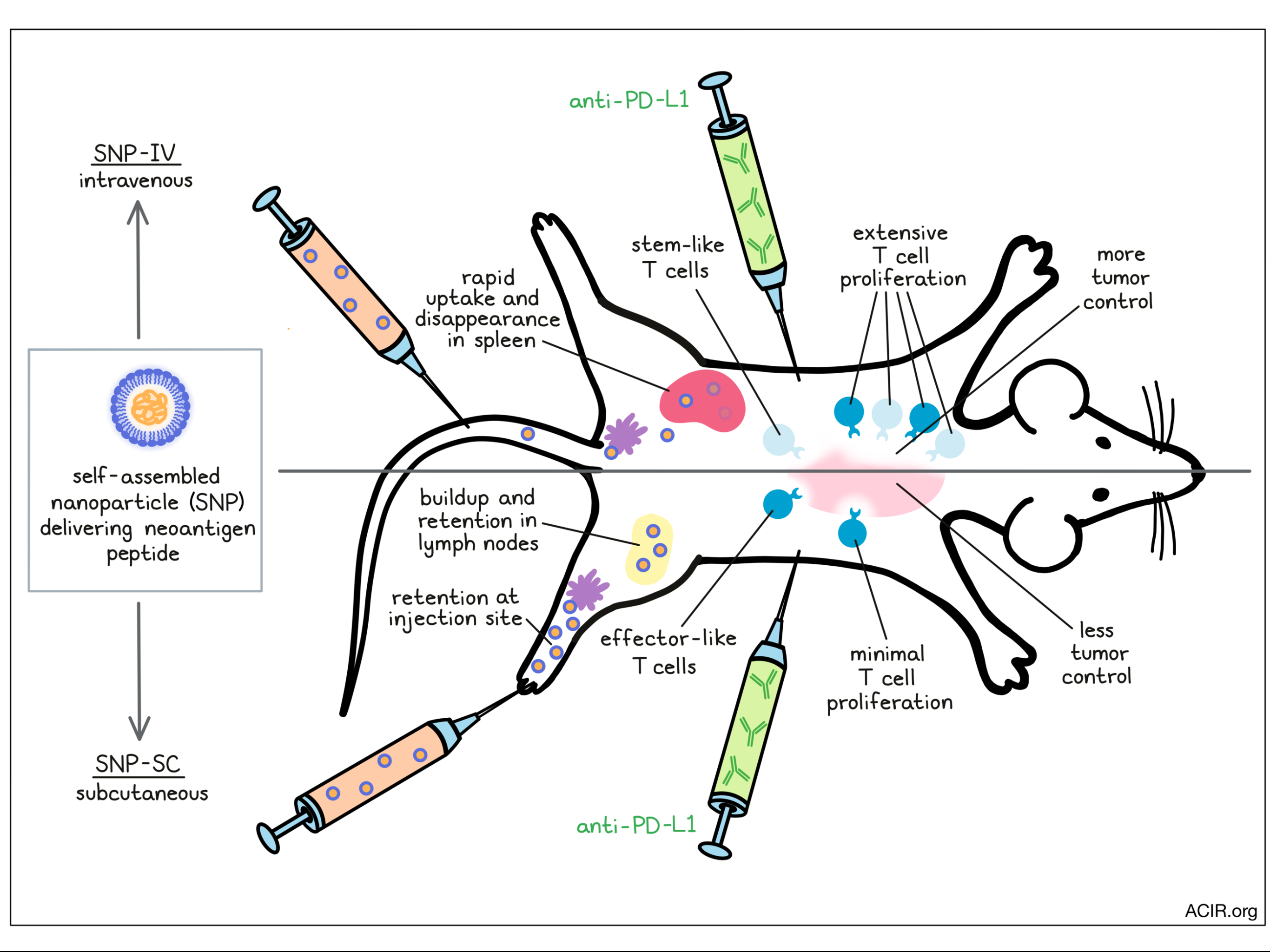
To address this hypothesis, Baharom et al. utilized a vaccine platform previously shown to generate robust neoantigen-specific T cell responses [1]. To deliver peptides with diverse physicochemical properties, charge-modified neoantigen peptides were linked to a hydrophobic block bearing a TLR7/8 agonist and self-assembled into 20-50nm nanoparticles (SNP). The authors vaccinated mice with SNP delivering Reps1 peptide, an MC38 neoantigen, either intravenously (SNP-IV) or subcutaneously (SNP-SC). Notably, at comparable, 8 nmol doses, SNP-SC induced 20-fold greater Reps1-tetramer+ CD8+ T cells than SNP-IV. Increasing the vaccine dose increased the tetramer proportion in either route. To compare the antitumor efficacy of each strategy, the researchers prophylactically vaccinated mice before MC38 tumor challenge and treatment with anti-PD-L1. Interestingly, while SNP-SC more effectively controlled tumors than SNP-IV, only SNP-IV showed a combinatorial effect with anti-PD-L1 to extend mouse survival. This result suggested that despite inducing fewer antigen-specific T cells, SNP-IV may generate T cells with a more favorable phenotype for anti-PD-L1 therapy.
The researchers next considered what phenotypic differences exist between vaccine-induced T cells from either route. Tetramer+ CD8+ T cells in the blood of SNP-SC-vaccinated mice expressed primarily a short-lived effector phenotype (KLRG1+CD127-), whereas SNP-IV induced markers of memory precursor cells (KLRG1-CD127+). In fact, the frequency of memory precursor cells correlated negatively with the magnitude of overall tetramer response. In mouse spleens, SNP-IV increased the proportion of CD8+ T cells expressing TCF1, a marker associated with stemness and immune checkpoint blockade response in humans, relative to SNP-SC. Indeed, these TCF1+ CD8+ T cells expressed CD127, EOMES, and CXCR3, indicating a memory or stem-like quality. This effect was antigen-independent, as replacing Reps1 with E7, ovalbumin, or Trp1 peptides recapitulated these results.
To further scrutinize these phenotypes, Baharom et al. performed single-cell RNA sequencing on tetramer+ CD8+ T cells sorted from spleens four weeks after the start of a prime/boost vaccination. Cells derived from SNP-IV vaccination associated primarily with stem-like cell clusters (expressing Tcf7, Tox, Slamf6, Xcl1, Eomes), while cells induced by SNP-SC clustered with effector-like clusters (expressing Gzma, Gzmb, Klrg1, Runx3). Pseudotime trajectory analysis pointed to a developmental pathway from naive to stem-like to effector-like cells, with SNP-IV/SC induced cells at the expected corresponding points along this trajectory. Importantly, the analyzed T cells from SNP-IV-vaccinated mice matched previous stem-like gene signatures from chronic LCMV infection, while SNP-SC induced T cells expressing effector-associated genes found in acute LCMV.
The researchers next compared the two SNP administration routes in a therapeutic prime-boost model along with anti-PD-L1. In this experiment, the SNP-IV dose was increased such that the overall magnitude of the antigen-specific T cell response was consistent with SNP-SC. Strikingly, despite the same number of Reps1-specific T cells between the two groups, SNP-IV was more effective in controlling tumor growth and extending mouse survival. The full regime of a vaccine prime, boost, and anti-PD-L1 was most effective and resulted in the highest frequency of antigen-specific T cells in the spleen, compared to a vaccine prime alone, prime + anti-PD-L1, or prime + boost without anti-PD-L1. As previously seen, SNP-IV induced primarily stem-like CD8+ T cells (~85% TCF1+GzmB- versus ~35% with SNP-SC). Anti-PD-L1 treatment following SNP-IV prime/boost expanded stem-like T cells in the spleen, but also effector T cells, suggesting a capacity for both self-renewal and differentiation.
Finally, the team examined the antigen presentation process associated with either delivery route. Fluorescently labeled SNP-SC was retained at the injection site, distributing to the draining lymph node (LN) subcapsular sinus over 24h, and to the T cell zone over the following two weeks. In contrast, SNP-IV distributed systemically and was detected within spleens at 6-24h, and minimally at later timepoints. Flow cytometric analysis found SNP-IV and SNP-SC taken up by both conventional DC1s (cDC1s) and monocyte-derived DCs (moDCs), although with different kinetics: SNP-SC uptake and DC activation were detected in LNs up to three weeks after vaccination, while SNP-IV uptake peaked in the spleen at six hours, and DC activation declined rapidly beyond day 2. Similarly, while both SNP-SC/IV induced cytokines IL-12 and IFNα, SNP-SC significantly prolonged their expression. In both delivery routes, deficiency in Batf3, Ccr2, Ifnar, or Tlr7, but surprisingly not IL-12b, attenuated the magnitude of vaccine T cell responses, indicating that cDC1s and moDCs are both essential, along with IFNα and TLR7 signaling. Deficiency of IL-12b also did not impact the frequencies of TCF1-expressing cells in SNP-SC vaccinated mice, indicating that the extended IL-12 expression was not responsible for the overall lower frequency of TCF1+ cells. Questioning whether antigen retention may also differ between the two routes, the researchers vaccinated mice, then transferred OT1 T cells at several timepoints over the following week. OT1 cells proliferated best when delivered 1 day following SNP-IV vaccination, compared to 7 days following SNP-SC vaccination, implying that SNP-SC indeed maintains antigen presentation at later timepoints, potentially due to slower antigen release from the subcutaneous injection site depot.
In conclusion, Baharom et al. demonstrated that a design parameter as simple as the delivery route of a nanoparticle vaccine had significant implications for the strength, phenotype, and efficacy of the T cell response. Furthermore, the suggestion that a reduced duration of antigen presentation may be more amenable to the formation of stem-like antigen-specific CD8+ T cells has important implications for vaccine delivery and would provide strategies to improve immune checkpoint blockade therapies.
Write-up by Alex Najibi, image by Lauren Hitchings
Reference:
[1] Lynn, G.M., Sedlik, C., et al. Peptide–TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nature Biotechnology 2020.
Meet the researcher
This week, first co-author Faezzah Baharom answered our questions.

What prompted you to tackle this research question?
Although initial studies on personalized cancer vaccines have been promising, the magnitude of CD8+ T cell responses have been limited. We designed a vaccine platform that could overcome the issues of efficient peptide uptake by dendritic cells. However, despite getting high magnitude responses by the conventional route of administration (e.g. subcutaneous or intramuscular), we could not demonstrate efficacy in therapeutic tumor models. As the Seder lab had prior experience in successfully delivering vaccines intravenously (malaria and TB vaccines), we tried the same approach with our cancer vaccine, and indeed it did improve antitumor efficacy!
What was the most surprising finding of this study for you?
The importance of CD8+ T cell quality. Despite generating similar numbers of neoantigen-specific CD8+ T cells after SC or IV vaccination, the cells were not equally capable of clearing tumors. We now know that the duration of antigen presentation is important in determining whether a CD8+ T cell remains stem-like or becomes terminally differentiated.
What was the coolest thing you’ve learned (about) recently outside of work?
I visited a lovely farm in rural Virginia recently and learned about sustainable farming. The basic principle of Polyface farms is to observe and emulate the animals’ behavior in nature. As an example, they established a rotational grazing system where the chickens follow the cows. This helps to improve fertilization of pastures, as the chickens spread the natural fertilizer left behind by cows, thus restoring the quality of the soil for future grazing.



