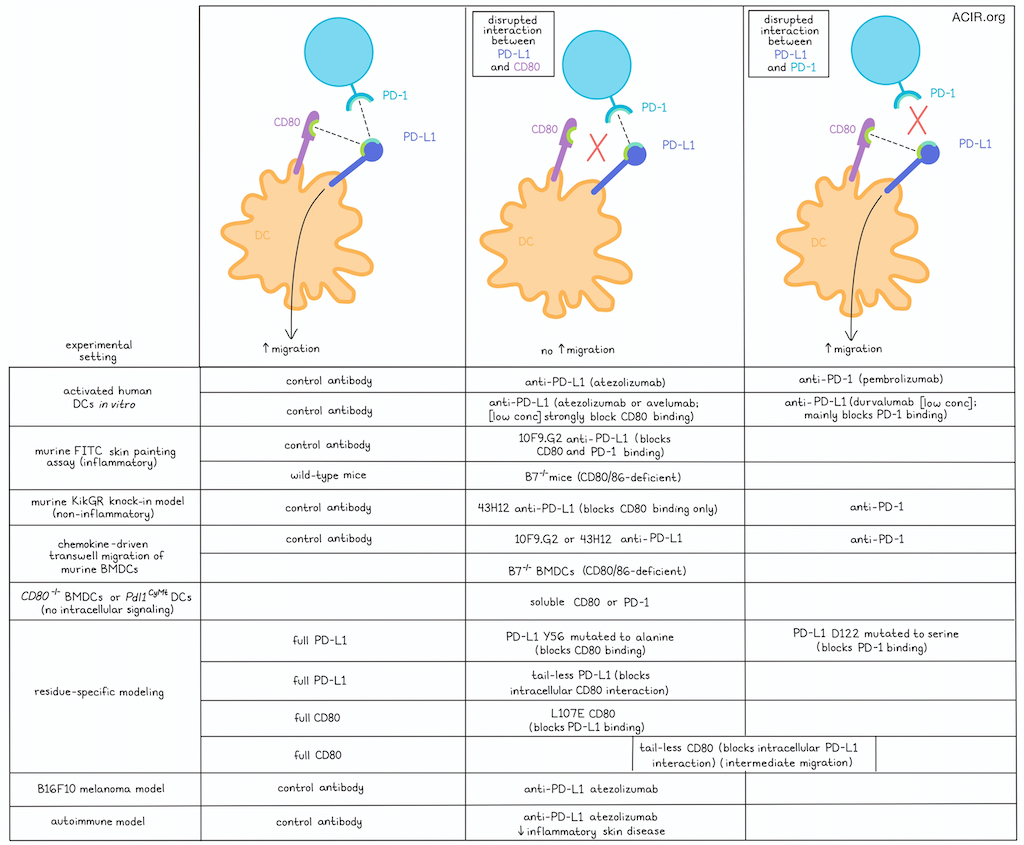
Research has shown that PD-L1 expression on dendritic cells (DCs) is essential for intracellular signaling that plays a role in chemokine-mediated DC migration. In a recent paper in Science Advances, Kantheti et al. investigated whether the extracellular interaction of PD-L1 on DCs with PD-1 or with CD80 in cis or in trans plays a role in this process.
The researchers first converted human CD14+ cells isolated from the peripheral blood into DCs using IL-4 and GM-CSF. These DCs showed chemokine-directed migratory behavior when activated with LPS. Blockade of PD-1 in this culture system by pembrolizumab did not affect this migration, but PD-L1 blockade by atezolizumab significantly reduced migration.
To investigate the effects of anti-PD-L1 antibodies on DC migration, the researchers then assessed their binding affinity and the amino acid residues masked by the antibody. The three currently approved clinical anti-PD-L1 antibodies were investigated for this: atezolizumab, durvalumab, and avelumab. The binding affinities of atezolizumab and durvalumab are similar, while avelumab has a stronger binding affinity. Testing these antibodies in the migration model showed that the antibody with the lower affinity, atezolizumab, inhibited migration most. However, all antibodies had similar inhibitory effects at high doses, consistent with fully saturating PD-L1 on the DCs. Moving to assessment of the amino acids masked by the antibodies, atezolizumab covered most residues in the CD80 binding domain of PD-L1 and formed a polar interaction with tyrosine-56 on PD-L1. Avelumab also formed a biochemical interaction with tyrosine-56, and while durvalumab, surprisingly, did not mask any of the known PD-L1 residues necessary for CD80 binding, it bound with residues essential for PD-1 binding.
Moving to murine models, Kantheti et al. investigated whether the interaction of PD-L1 with PD-1 or CD80 is important for in vivo DC migration from the skin to the draining lymph node (dLN). An FITC skin painting assay was used to mark dermal DCs to allow tracking to the dLN. In mice that had received an injection of polyI:C, treatment with the anti-PD-L1 clone 10F9.G2, which has similar PD-L1 binding as atezolizumab, resulted in fewer FITC+ cDC1s and cDC2s in the skin dLNs, compared to isotype control antibody. Moreover, the cDCs in the anti-PD-L1-treated mice had lower PD-L1 and CD80 expression. Further, in B7-/- mice, which lack CD80 and CD86, fewer DCs were detected in the dLNs compared to WT mice after polyI:C injection.
As FITC skin painting is an inflammatory model, the researchers then moved to the KikGR knock-in model, in which exposure to violet light causes the conversion of Kikume green fluorescent protein to red fluorescence, a non-inflammatory alteration. To further assess whether PD-L1:CD80 interactions are needed for DC migration, another anti-PD-L1 antibody clone was used, 43H12, which only inhibits the interaction between PD-L1 and CD80, not between PD-L1 and PD-1. Treatment with this antibody in this model resulted in fewer cDC1s and cDC2s in the dLN. When an anti-PD-1 antibody was used, no differences in cDC1s or cDC2s were observed in the dLN.
The researchers then aimed to establish whether the differences in DC migration were specific to chemokine migration. Using a transwell system and bone marrow-derived DCs (BMDCs) from mice, cells treated with isotype control or anti-PD-1 antibody migrated effectively when activated by LPS, while cells treated with two anti-PD-L1 antibodies (10F9G.2 and 43H12), or B7-/- BMDCs did not migrate well. Further, Cd80-/- BMDCs, which still have CD86, also did not migrate well, suggesting the loss of CD80 causes reduced migration.
Next, experiments assessed whether DC migration depends on cis or trans PD-L1:CD80 interactions. Adding soluble CD80 or PD-1 to cultures with Cd80-/- BMDCs did not improve migration. Previously, the researchers had shown that when PD-L1 cytoplasmic residues T277, S278, and S279 are converted to alanine, DC migration is impaired. Culturing these mutated Pdl1CyMt DCs in the presence of exogenous soluble CD80 or PD-1 also did not rescue migration. These data suggest that PD-L1 signaling-induced DC migration results from cis interactions with CD80.
To determine which residues were important for DC migration, murine PD-L1 Y56 was mutated to alanine, and D122 to serine, to prevent cis PD-L1 binding with CD80 or PD-1, respectively. Further, the cytoplasmic tail of PD-L1 at residue 275 was deleted to abolish CD80:PD-L1 intracellular interactions, which had also been shown to reduce migration. Pdl1-/- BMDCs reconstituted with full-length PD-L1 or D122S PD-L1 migrated more than BMDCs that expressed Y56A PD-L1 or tail-less PD-L1. Similarly, Cd80-/- BMDCs were reconstituted with full-length CD80, L107E CD80, and CD80 with residue 276 replaced with a stop codon (tail-less CD80). BMDCs reconstituted with full-length CD80 BMDCs had better migration than those expressing L107E, while BMDCs reconstituted with tailless CD80 had an intermediate migratory phenotype.
Moving to a tumor model, the researchers assessed whether defective DC migration can also be observed after atezolizumab treatment. In the B16F10 melanoma model, atezolizumab was injected together with a transfer of violet proliferation dye-labeled BMDCs. Fewer transferred DCs migrated to the dLN when the antibody was present, and the cells expressed lower levels of PD-L1 and CD80.
Finally, the researchers asked whether the inhibition of DC migration might limit inflammatory responses in autoimmune diseases. They assessed this in a murine model of psoriasis-like dermatitis induced by applying the TLR7 agonist imiquimod (IMQ) to the skin. In this model, intraperitoneal injection of an anti-PD-L1 antibody resulted in reduced migration of DCs to the skin dLNs, and the DCs had lower expression of PD-L1 and CD80. When IMQ was applied for 5 days, epidermal thickening was increased in the untreated mice. However, in the mice treated with the antibody, there was limited epidermal thickening, and fewer IL-17-producing CD4+ T cells were detected in the dLN.
Together, this study showed the roles PD-L1 and CD80 on DCs play in their chemokine-driven migration to the LNs upon activation, and how anti-PD-L1 antibodies can limit this migration. In inflammatory disease, this could have beneficial effects, while in cancer immunotherapy, it may explain the more limited responses seen for anti-PD-L1 as compared to anti-PD-1 treatment, as it may reduce the overall antitumor T cell response.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Uma Kantheti answered our questions.

What was the most surprising finding of this study for you?
The most fascinating result for me was how clinically available PD-L1 antibodies (atezolizumab, durvalumab, and avelumab) all had a crucial effect on human dendritic cell migration. Until then, we had used exclusively murine experimental models to assess how dendritic cells migrated, and this particular set of experiments was the first step for us to translate this finding into human biology. Given how critical dendritic cells are in antigen presentation, especially in a tumor setting, showing that PD-L1 antibodies have this effect on DC migration is something that we hope is considered in understanding patient outcomes to different immunotherapies.
What is the outlook?
Our lab is looking to take this research in the direction of further understanding how downstream T cell responses are affected by PD-L1 intracellular signaling within dendritic cells in the context of skin infection. We are currently investigating how skewing localization of dendritic cells between the skin and the lymph node can affect T cell-mediated immunity during cutaneous virus infection. Since virtually nothing is known about the consequences of PD-L1 signaling in skewing of memory T cell phenotypes, these future studies will also be highly impactful.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
I would tell myself to always go in the direction of where the science and the experimental data are taking you. Oftentimes, our initial hypotheses end up being totally wrong, which is okay, since the point of scientific experimentation is to further our understanding and to make breakthroughs in science and medicine. The unpredictability of what is not known is what makes being a scientist so exciting!



