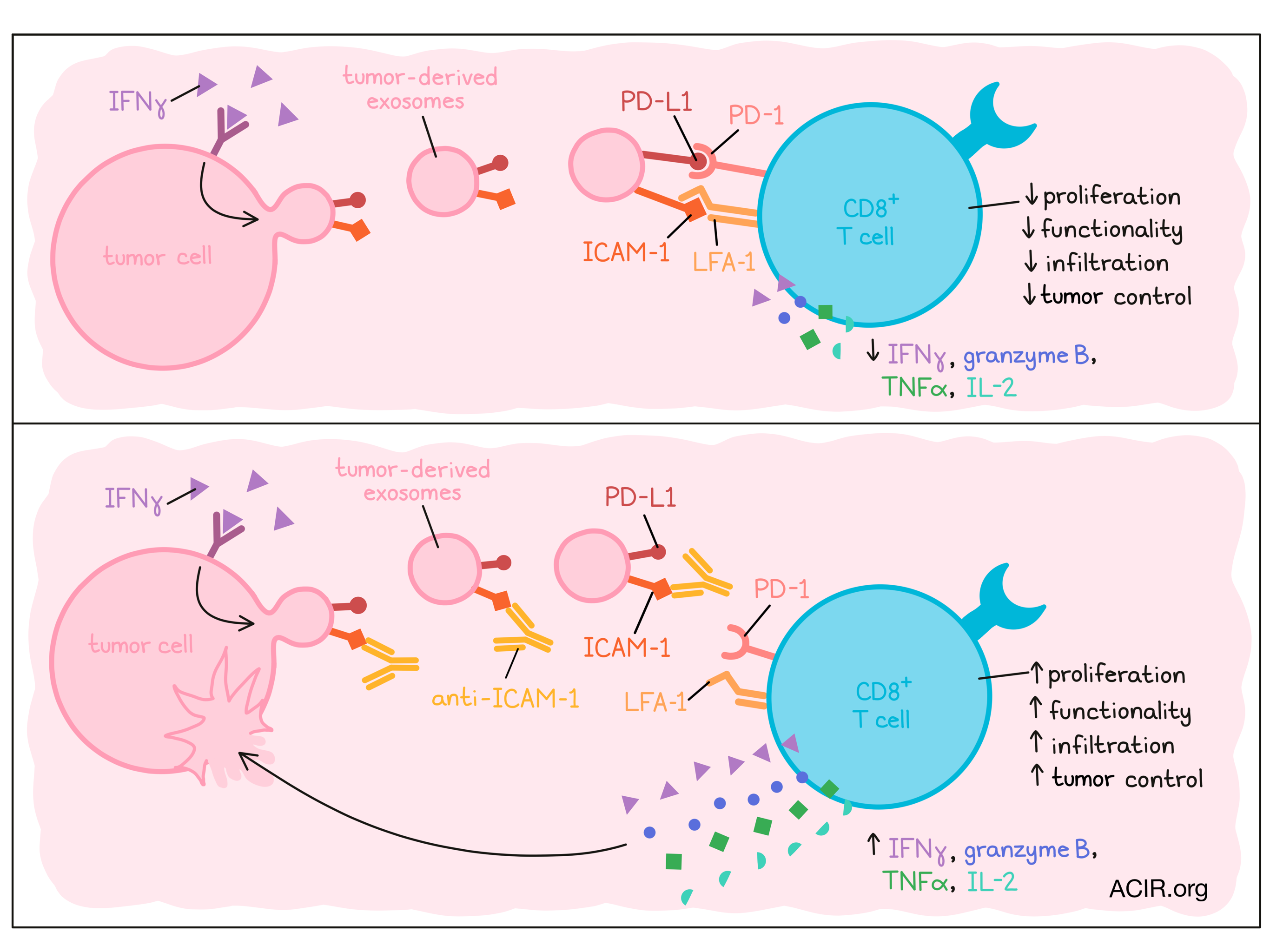
Tumors employ diverse tactics to suppress CD8+ T cell immunity, one of which is secreting tumor-derived extracellular vesicles (TEVs) that express PD-L1. However, exactly how TEV PD-L1 interacts with CD8+ T cells remains unknown. Recently reported in Developmental Cell, Zhang and Zhong et al. uncovered a novel role of the adhesion molecule ICAM-1 on TEVs for dampening CD8+ T cell activity.
To begin, Zhang, Zhong, and the team characterized ICAM-1 expression on TEVs from human melanoma cell lines. They found that ICAM-1 was specifically enriched in melanoma-derived exosomes (as opposed to microvesicles) and upregulated along with PD-L1 in response to IFNγ exposure. Initially observed through Western blotting, this result was recapitulated using an ELISA the authors developed in house to characterize TEV-expressed proteins. Further confirming that ICAM-1 is expressed in TEVs, the authors found that ICAM-1 co-localized and co-immunoprecipitated with Hrs, a critical component of the EV loading and release pathway, within melanoma cells. shRNA knockdown of Hrs or Rab27a, another regulator of exosome release, reduced ICAM-1 levels in secreted exosomes. Altogether, these results demonstrate the presence of ICAM-1 on TEVs.
Next, the researchers questioned whether ICAM-1 colocalizes with PD-L1 on TEVs. Using immuno-electron microscopy, they visualized expression of these molecules on the same exosomes. Consistent with their prior findings, the fraction of ICAM-1+PD-L1+ exosomes increased with IFNγ treatment. Furthermore, depletion of ICAM-1+ exosomes using anti-ICAM-1 Dynabeads reduced PD-L1 levels compared to non-depleted exosomes and the bead-enriched exosomes. Interestingly, ELISA analysis found higher ICAM-1 expression on PD-L1+ exosomes isolated from the plasma of metastatic melanoma patients compared to those isolated from healthy patients. Thus, ICAM-1 and PD-L1 can be co-expressed on exosomes and may play supporting roles in tumor progression.
The team next characterized TEV interactions with CD8+ T cells. CFSE-stained exosomes could bind to CD8+ T cells, and significantly better when the cells were pre-stimulated. Blocking ICAM-1, either through an anti-ICAM-1 mAb or shRNA-mediated ICAM-1 knockdown, diminished this binding. To determine which receptor could be binding ICAM-1, the researchers targeted LFA-1 or Mac-1 – known ICAM-1 binding partners. Blocking LFA-1, but not Mac-1 reduced TEV binding of CD8+ T cells, and accordingly, LFA-1 was upregulated on CD8+ T cells after stimulation. These results indicated that ICAM-1 on TEVs may bind CD8+ T cells, specifically through LFA-1, which is upregulated by T cells upon their activation.
The authors then questioned whether ICAM-1 on TEVs plays a role in CD8+ T cell suppression. First, they confirmed that exposure to melanoma TEVs reduced CD8+ T cell functionality. Indeed, NFAT activation by a Jurkat reporter cell line; T cell proliferation; Ki67 and GzmB expression; and IL-2, TNFα, and IFNγ secretion were all diminished upon incubation of T cells with TEVs. Importantly, blockade of ICAM-1 (either through an anti-ICAM-1 mAb or shRNA knockdown) on the TEVs restored these functional parameters. These results were not unique to melanoma; colon and lung cancer TEVs had similar suppressive effects that were reversed by ICAM-1 blockade. PD-L1 was necessary for this suppression, but not involved in T cell binding, as, compared to wild-type melanoma TEVs, PD-L1-/- TEVs bound T cells similarly, but had reduced suppressive capacity.
Next, Zhang and Zhong et al. explored their findings in vivo using YUMM1.7 melanoma tumors. Tumor-bearing mice injected intravenously with TEVs showed accelerated tumor growth, decreased CD8+ T cell infiltration, and reduced Ki67 and GzmB expression among intratumoral PD-1+ CD8+ T cells, compared to control mice that were not given TEVs. However, these effects were lost when the infused exosomes were derived from ICAM-1-/- YUMM1.7 cells or wild-type exosomes pre-treated with anti-ICAM-1 mAb. These findings were mirrored in the MC38 colon cancer model. These in vivo results complemented the team’s prior studies, further indicating a role of ICAM-1 in abetting TEV-mediated immunosuppression.
Finally, the researchers developed an exosome–cell receptor binding assay to confirm the role of ICAM-1 in T cell suppression through PD-L1. They leveraged SrtA, a bacterial protein used in labeling studies that can transfer a biotin-labeled peptide motif to a target bearing a GlyGlyGlyGlyGly (G5) tag. The authors engineered melanoma cells to express SrtA fused to PD-L1, and Jurkat T cells to express the G5 tag fused to PD-1. In theory, the melanoma cells would produce TEVs expressing the PD-L1–SrtA fusion, which would be capable of labeling G5–PD-1 on the T cells when brought in proximity. The labeling performed as expected when the engineered T cells and TEVs were co-incubated, confirming that PD-L1 on the TEVs and PD-1 on the T cells do come in contact. Importantly, pre-treatment of TEVs with anti-ICAM-1 mAb or ICAM-1 knockdown reduced this labeling, suggesting that ICAM-1 may be required for optimal PD-L1–PD-1 binding between TEVs and T cells.
In summary, Zhang and Zhong et al. found a critical role of the adhesion molecule ICAM-1 on tumor cell-derived exosomes in enabling PD-L1-mediated immunosuppression of CD8+ T cells. Further understanding mechanisms of tumor resistance to endogenous immunity or immunotherapy could support advances in immune checkpoint blockade and other cancer therapies.
Write-up by Alex Najibi, image by Lauren Hitchings



