Despite the important role that Type I (IFNα and β) and Type II (IFNγ) interferons have in antitumor immunity, both by upregulating HLA in cancer cells and activating T cells, prolonged IFN signaling can be pro-tumorigenic and has been associated with immunotherapy resistance. Therefore, Qiu and Xu et al. investigated whether interfering with IFN-I signaling could improve response to immune checkpoint blockade (ICB), and assessed its impact on adaptive immune responses. Their results were recently published in Nature Cancer.
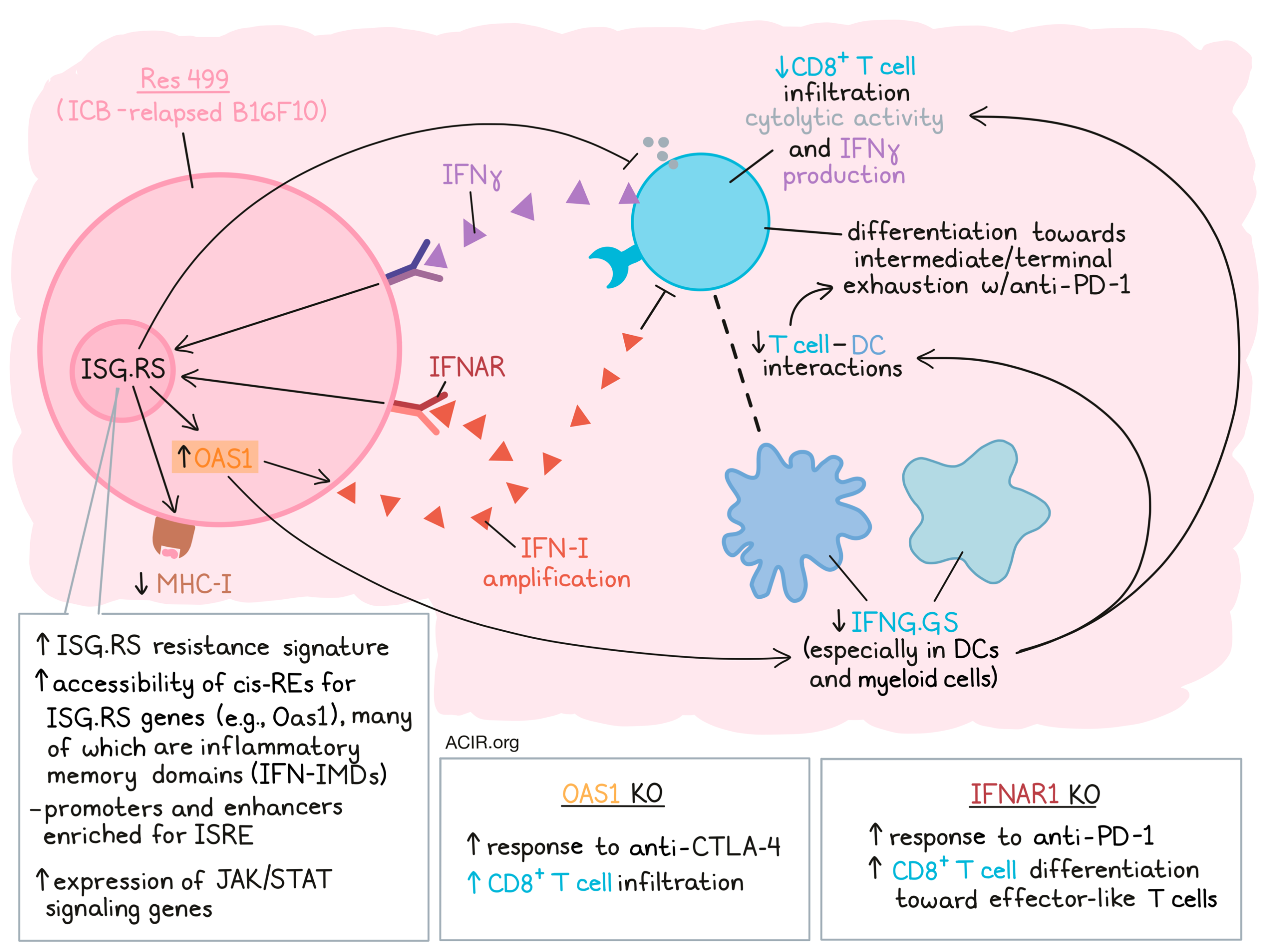
The researchers had previously identified two subsets of IFN-stimulated genes (ISGs); ISG.RS (resistance signature), which is associated with IFN-I and is predominantly expressed by cancer cells, and IFNG.GS (gene signature), which predicts clinical response to ICB in melanoma and is mainly expressed by immune cells. To assess whether these ISGs were associated with ICB response in other cancer types, The Cancer Genome Atlas (TCGA) was interrogated for gene expression data. Increases in IFNG.GS expression were associated with elevated CD8+ T cell activity, but only when ISG.RS was expressed at low levels. Given that not all cancers developed high ISG.RS expression levels, the researchers hypothesized this might be due to the epigenetic state of the cancer cells.
To investigate this, paired RNAseq and ATACseq data were analyzed in the context of TCGA data to model gene expression, focusing on cancer cell-specific chromatin accessibility. This allowed the prediction of putative cis-regulatory elements (cis-REs) for the ISG.RS genes. The expression of most genes in the ISG.RS subset could be explained by chromatin accessibility, suggestive of epigenetic regulation. This chromatin accessibility of ISG.RS cis-REs was also negatively associated with CD8+ cytolytic activity. One ISG.RS gene with evidence of strong epigenetic regulation was OAS1. The cis-REs linked to OAS1 were inaccessible in tumors with high CD8+ T cell activity, but accessible in tumors with low CD8+ T cell activity.
To further investigate how the cancer cell chromatin landscape promotes high ISG.RS expression and ICB resistance, the researchers used a mouse model derived from an ICB-relapsed B16F10 melanoma tumor (Res 499), which has high expression of ISG.RS genes. Res 499 cells had undergone a striking remodeling of the chromatin landscape as compared to B16F10 cells. In addition, the expression of Oas1 was found to be regulated by cis-REs in these cells.
Next, the researchers used the mouse datasets to define an in vivo catalog of promoters and enhancers that were activated, deactivated, or constitutive in Res 499 tumors compared to B16F10. The enhancers and promoters activated in Res 499 cells were enriched for motifs associated with the archetypal IRF/2 IFN-stimulated response elements (ISRE) predicted to accommodate STAT1-STAT2 heterodimers and IFN regulatory factor (IRF) family transcription factors. In addition, Res 499 tumors were enriched for genes associated with the JAK-STAT signaling pathway.
To assess whether the Res 499 activated enhancers were linked to chronic IFNγ signaling, B16F10 cells were stimulated with IFNγ for 6 hours (acute) or 3.5 weeks (chronic) in vitro before implantation into mice. Established tumors from cells that had been chronically stimulated had increased chromatin accessibility at approximately a quarter of Res 499 activated enhancers, and these enhancers were also enriched with IRF/2 motifs.
To uncouple epigenetic changes due to ongoing IFN signaling from those maintained after signaling stops (“inflammatory memory domains” [IFN-IMD]), the researchers used Stat1 knockout in Res 499 or B16F10 tumors before implantation to identify IFN-IMDs that maintain chromatin accessibility. Approximately 21% of these IFN-IMDs overlapped with Res 499 activated enhancers, and were not just enriched for IFN signaling transcription factors (TFs), but also those belonging to other cytokine families. To determine putative inflammatory memory genes, gene expression was linked to IFN-IMD chromatin accessibility after chronic IFNγ stimulation. A total of 611 genes were detected, which were also elevated in Res 499 tumors, and most ISG.RS genes were included in these inflammatory memory genes.
The researchers then assessed whether IFN-IMDs enabled cancer cells to amplify IFN-I signaling, sustain high ISG.RS expression, and maintain immune dysfunction. Given its high expression in Res 499, Oas1 was deleted by CRISPR to assess these effects. After stimulation with polyI:C, the resistant Res 499 cells produced higher levels of IFN-I and ISGs than non-resistant parental B16F10 cells, an effect almost completely abrogated in cells with the Oas1 KO. These results suggested the IFN-I signaling amplification occurs through OAS1 signaling in resistant cells.
Oas1 KO in ICB-resistant Res 499 recovered the response to anti-CTLA-4 and was associated with high CD8+ T cell infiltration. To determine how inhibition of cancer cell IFN-I signaling helps restore response to ICB, the researchers investigated Ifnar1 KO in the Res 499 model. This KO improved responses to anti-PD-1, increased cancer cell MHC-I expression, and decreased the expression of genes previously associated with anti-PD-1 resistance. These effects were also seen in co-implantation studies (Ifnar1 KO Res 499) and when only a limited number of cancer cells had the Ifnar1 KO, suggesting restoration of a missing function in Res 499 by Ifnar1 KO.
Looking at immune populations in Res 499 tumors, the researchers found that Ifnar1 KO in cancer cells increased immune cell expression of IFNG.GS, in particular in DCs and myeloid cells. This resulted in increased Ifng expression by NK and CD8+ T cells, particularly in effector-like and exhausted subsets. Cell–cell interaction in silico assessments revealed that in Res 499 tumors without IFNAR and treated with anti-PD-1, the strongest interactions were between CD8+ T cells and the DC subset DC3, with increased communication probability between multiple co-stimulatory receptor–ligand pairs between these cell types.
Finally, the researchers assessed the impact of these enhanced interactions on CD8+ T cell fate. Res 499 tumors mostly contained progenitor-exhausted T cells that evolved to intermediate and terminal exhausted subtypes upon treatment with anti-PD-1. However, Ifnar1 KO and anti-PD-1 treatment resulted in an expansion of effector-like T cells, an effect that was found to be TCR-dependent.
In sum, Qiu and Xuet al. show that IFN signaling in cancer cells, a result of T cell-mediated IFN production that ultimately leads to T cell suppression, establishes an epigenetic state that maintains expression of genes carrying that suppressive capability in a feedforward loop when IFN signaling is lowered due to suppression of the T cells. These results suggest that interfering with IFN-I signaling in cancer cells may improve ICB responses in those with acquired resistance, and may therefore be of interest to combine with ICB in clinical studies.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first authors Jingya Qiu and Bihui Xu answered our questions.

What was the most surprising finding of this study for you?
We have known for many years that prolonged interferon (IFN) signaling and elevated expression of interferon-stimulated genes (ISGs) in cancer cells can promote resistance to immune checkpoint blockade (ICB). Therefore, after we realized that chronic IFN was also linked to epigenetic changes in resistant cancer cells, we were surprised to find that preventing IFN signaling did not reverse these epigenetic changes like it did ISG expression and ICB resistance. This unexpected result was what motivated us to explore how and why epigenetic changes persist, and to understand its significance for ICB resistance in both mice and humans.
What is the outlook?
Inflammatory memory, in which cells “remember” inflammatory experiences in its chromatin to adapt to or respond more robustly to subsequent stimuli, has previously been characterized as a facilitator of trained immunity and enhanced wound healing in immune cells and epithelial cells, respectively. Our study suggests that in cancer, inflammatory memory is also involved in acquired ICB resistance, driven by chronic IFN signaling. Our work highlights how cancer cell epigenomes may evolve because of chronic inflammation and the consequences for immunotherapy resistance. We think our findings provide insight into why optimizing timing and duration of cytokine and inflammatory signaling is an important aspect in designing immunotherapy strategies. Next steps include investigating the therapeutic significance of IFN-driven inflammatory memory, and identifying ways to prevent or reverse it, with the goal of improving immunotherapy efficacy.
What was the coolest thing you’ve learned (about) recently outside of work?
BX: I recently finished the book “How to win friends and influence people”, and am working through “How to talk so little kids will listen”. I am surprised to find that many principles taught by the two books are similar. I guess it should not come as a surprise, since after all, little kids are people too and deserve the same respect as adults do. I am also learning that managing a two-year-old at home is no less work than managing adults at work!
JQ: I’m excited to re-join the world after just defending my thesis… hobbies are back on the table again!



