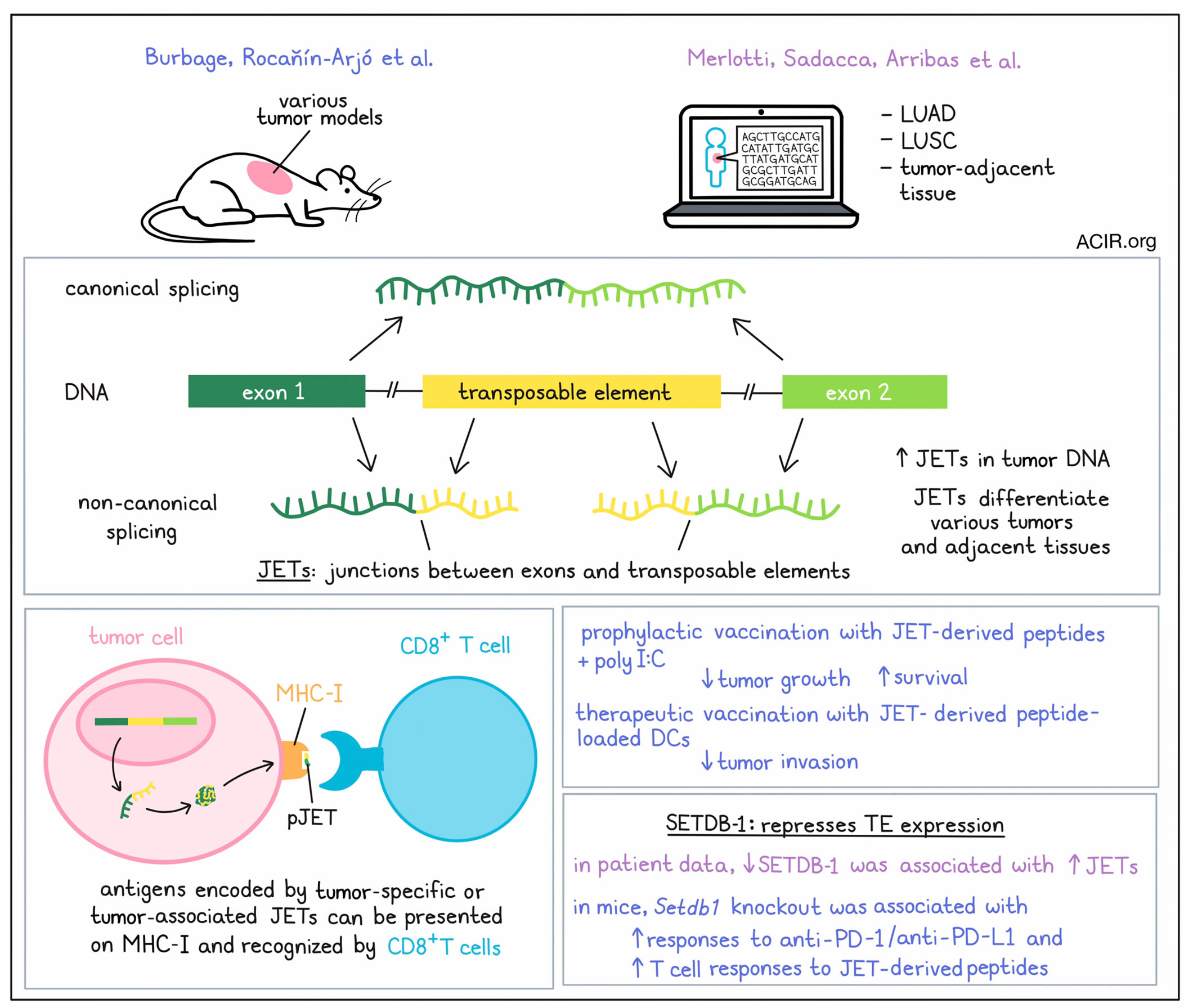
Epigenetic alterations, such as the derepression of transposable elements (TEs), are common events in cancer. Two recent papers from Amigorena and colleagues published in Science Immunology investigated whether non-canonical splice junctions between exons and TEs may serve as a source of tumor antigens in murine models and patient samples.
To identify non-canonical splicing events between coding exons and TEs, a pipeline was developed to detect splicing RNAseq reads that map partially to a coding exon and partially to a TE, called JETs (junctions between exons and TEs). Burbage, Rocañín-Arjó et al. used RNAseq datasets from several mouse tumor cell lines and identified approximately 400 JETs per tumor cell type. Comparing JET expression between various tumor cell lines, as well as healthy tissues, showed that JET expression could differentiate these tissues and tumors in various clustering analyses. Primary melanocytes and B16OVA cells could also be separated based on their JETs patterns. Therefore, JETs were expressed in healthy and tumoral cells, and tumor cells had specific JET sets that differentiated them from healthy cells.
Similarly, Merlotti, Sadacca, Arribas, et al. searched for JETs in lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and tumor-adjacent normal tissue from TCGA, and found that JETs were also expressed in both healthy and tumoral tissues in humans. JETs were detected in all samples, with higher numbers in the tumor samples than in the juxta-tumor samples. Clustering analyses also showed that JETs could separate juxta-tumor samples by tissue type, as well as separate between cancer and healthy cells, and between LUAD and LUSC samples.
The researchers next assessed whether the JETs overexpressed in tumors could represent a source of tumor antigens. 53% of LUAD and 59% of LUSC JETs were not present in the juxta-tumor samples. Overall, most JETs were expressed at a low frequency, while the most recurrent JETs were expressed in both tumor and healthy tissue. However, a small subset of JETs was recurrent and only present in tumors – these were referred to as tumor-specific JETs (tsJETs). A second group was considered tumor-associated JETs (taJETs), because they were present in a high percentage of tumors and a low percentage of healthy tissue. Even when the researchers expanded datasets with more healthy tissue samples, many of the tsJETs and taJETs were not present or very rarely expressed in normal tissue.
To understand whether peptides encoded in JETs were presented on MHC-I, Burbage, Rocañín-Arjó et al. conducted an in silico analysis of all potential peptides encoded by the murine JETs to search immunopeptidomic datasets for JET peptides. Indeed, 19 peptides were identified, suggesting that JETs generated peptides that could be a source of tumor antigens. Additional similar MHC-binding peptides were identified by predicting binding using netMHC. A set of identified and predicted peptides were synthesized, and RMA-S cells, which lack the transporter associated with antigen processing (TAP), were used to assess the binding to MHC-I. Most synthetic peptides could stabilize MHC-I expression.
Next, the researchers aimed to evaluate whether these MHC-I-presented murine peptides could be recognized by the immune system by stimulating splenocytes from mice bearing MC38 tumors and control mice with JET-derived peptides. T cell responses were detected against some of the JET-derived peptides in T cells from tumor-bearing mice, while very few were detected in healthy mice. These T cell responses were also detected when the less immunogenic tumor, B16OVA was used. These results were confirmed using two-color tetramer staining of T cells from lymph nodes and tumors.
In the human study, Merlotti, Sadacca, Arribas, et al. also assessed whether JET peptides could be presented by HLA-I molecules in tumors. In silico analysis similarly revealed 114 peptides that were potentially encoded by tsJETs, which they named pJETs. pJETs had expected HLA-I-binding motifs and may represent a source of HLA-I-presented peptides.
To assess whether the pJETs were immunogenic, the researchers performed in vitro experiments using pJETs that were in silico-predicted to bind HLA-A*02:01 and were derived from recurrent tsJETs. The 31 most recurrent tsJETs were selected, which coded for 59 unique peptide sequences; 47 synthetic peptides were produced. Then, an in vitro peptide-HLA-I complex formation assay was performed to confirm the binding of 29 of these peptides to HLA-A*02:01. About half of patients in the LUAD and LUSC cohorts expressed at least one tsJET from this panel, and at least one-third of these were tumor-specific. The immunogenicity of these 29 JETs was assessed by coculturing peptide-loaded HLA-A*02:01 monocyte-derived DCs with autologous CD4+ and CD8+ T cells from healthy donors. pJET-specific tetramer+ populations were found for most tested pJETs, and all donors were positive for at least one pJET. Reexpressing the sequenced TCRs of these T cells in TCR-negative Jurkat triple-reporter cells showed specificity in killing target cells loaded with the pJET.
To investigate whether these pJET-specific CD8+ T cells could be found in patients, samples of five patients with primary, untreated LUAD were assessed. Several pJET tetramer+ CD8+ T cell populations could be detected and expanded in culture with the pJET mix and IL-2. JET-specific T cells for at least one of 29 tested JETs were found in three out of four patients. Those detected in the tumor had an effector/memory phenotype, while those found in blood and lymph nodes were variably of less differentiated or naive/central memory phenotypes.
Finally, Burbage, Rocañín-Arjó et al. assessed whether T cell responses against these tumor-specific antigens encoded by JETs could be targeted for therapeutic purposes in murine models. They first assessed whether T cell responses against JET-encoded antigens could inhibit tumor growth. C57BL6 mice were immunized with long peptides encompassing the predicted JET-encoded MHC-I-binding sequences, with polyI:C as an adjuvant. When draining lymph nodes were collected, vaccine-induced T cell responses against the JETs were detected. To assess whether these T cell responses could protect against tumor growth, immunized mice were challenged with B16OVA cells. Vaccination with three out of seven JETs reduced tumor growth and improved survival. In a therapeutic vaccination study, B16OVA cells were intravenously injected to establish lung tumors, after which mice were immunized with splenic DCs that were activated with LPS and loaded with peptides. The JET peptide tested decreased lung tumor invasion.
To assess how TE repression influenced JET expression, both research groups looked into the histone methyltransferase SETDB1, which is involved in the repression of TE expression. In human tumor samples, SETDB-1-low samples differentially expressed more JETs than SETDB-1-high samples, suggesting the TE derepression in tumors may influence JET expression. In the mouse study, WT mice were injected with Setdb1-KO B16OVA cells before immune checkpoint blockade (ICB). The tumors deficient in Setdb1 showed better responses to anti-PD-L1/PD-1 as compared to controls. To determine whether peptides derived from Setdb1-specific JETs could be targeted by the immune system during ICB treatment, mice with B16OVA control or Setdb1-deficient tumors were treated with anti-PD-L1. This revealed a robust induction of T cells that were specific to several Setdb1-regulated JET-derived peptides. Therefore, JETs induced by Setdb1 inactivation in tumor cells could be targeted by T cells during ICB treatment.
The data from these two studies suggest a new source of tumor antigens that can induce tumor-specific immune responses in both murine models and patient samples. The mouse studies suggest that these responses can be further enhanced by immunotherapy approaches, and future research should assess whether and how this can be applied in the clinic to increase responses to immunotherapy in patients.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first author on “Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer”, Yago Arribas, and co-first author on “Epigenetically controlled tumor antigens derived from splice junctions between exons and transposable elements”, Marianne Burbage answered our questions.

What was the most surprising finding of this study for you?
YA: We have described a subpopulation of alternative splicing events that can induce an antitumor immune responses. We could expect that these events are mistakes derived from transcriptional noise, however, it was surprising to see how recurrent they were across patients, and that they could be translated and presented on the tumor cell surfaces by the antigen presentation machinery.
MB: In this study, we analyzed non-canonical splice variants, including parts of transposable elements (TEs), that could be effectively targeted by the immune system in preclinical mouse models. One thing that was amazing for me was that once the pipeline was established, we were able to (rather) quickly identify peptides that were as efficient as classical model antigens.
What is the outlook?
YA: These studies contribute to the understanding of a large novel landscape of neoantigens that can be used to target tumors by the immune response. One of the next steps is to prove if they are useful targets for TCR-based therapies or vaccination approaches. We are currently working to further advance in their clinical potential.
MB: Our results showed that TE inclusion into non-canonical splice variants required a permissive epigenetic context. One key challenge arising from this study is to identify additional molecular players involved in this process. This will open new avenues to modulate tumor immunogenicity and improve immunotherapeutic protocols.
What was the coolest thing you’ve learned (about) recently outside of work?
YA: I have recently acquired an old and very beautiful bike, and I really enjoy cycling through the streets of Paris. It is a very special way of discovering new corners of the city where I had never been before.
MB: I have a small daughter currently (or soon to be) learning to speak, but it is baffling to see how she already has building blocks of logic at her own scale. It is a daily reminder that reality can be seen from different points of view.



