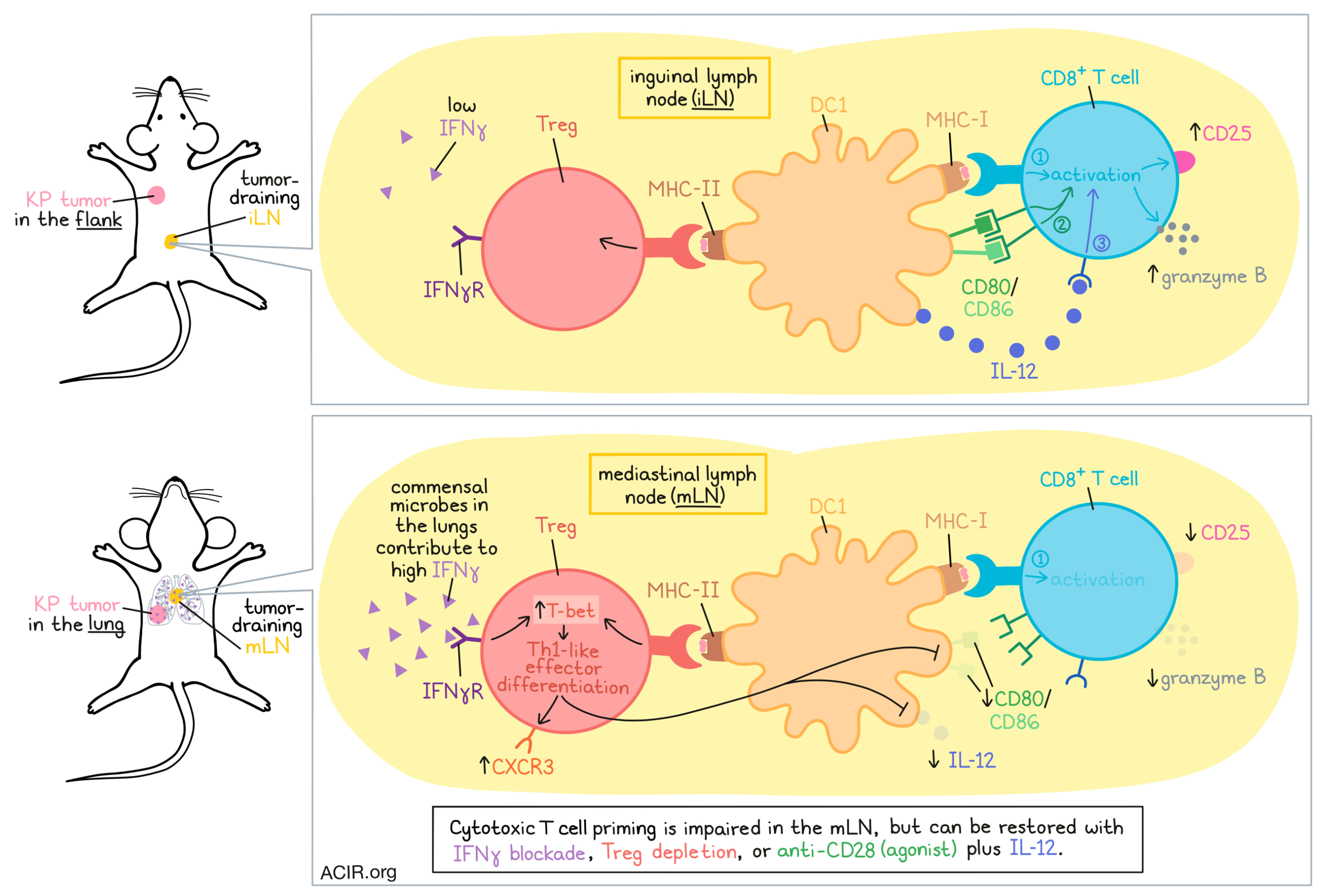
In an effort to better understand why mice mount weaker immune responses to tumors in the lungs than to those in the flank, Zagorulya et al. compared immune priming in their respective tumor-draining lymph nodes: the mediastinal lymph node (mLN) and the inguinal lymph node (iLN). As they unraveled this mechanism underlying these location-specific differences, the team identified a challenge to antitumor immunity that occurs before the cytotoxic T cell response even begins.
The researchers began by transplanting KrasG12DTrp53-/- (KP) lung adenocarcinoma tumors – engineered to express SIINFEKL fused to ZsGreen (ZsG) – either orthotopically into the lungs, or subcutaneously into the flanks of mice and compared T cell responses. While SIINFEKL-specific CD8+ T cells accumulated similarly in each LN, those from the mLN were less activated, expressing less CD25, granzyme B, and TIM3 than those primed in the iLN.
After determining that this differential effect was not likely to be due to TCR signal strength, the researchers turned their attention towards antigen-positive (ZsG+) cross-presenting DC1s, which were required for productive T cell priming in both tumor-draining lymph nodes (tdLNs). While DC1 abundance, maturation (MHC-II, CCR7), and signal 1 (antigen presentation) were similar between tdLNs, signal 2 (costimulation; CD80, CD86) and signal 3 (cytokines; IL-12) were impaired in DC1s from the mLN. This difference was not apparent in naive mice, nor in ex vivo priming studies, suggesting that this effect was not intrinsic to the DC1 or mLN, but rather, something associated with the tumor setting.
Zagorulya et al. found that depletion of Tregs in mice could rescue expression of CD25 and granzyme B on tumor-reactive T cells in both tdLNs, but that this effect was more pronounced in cells primed in the mLN. Transient Treg depletion also increased CD80 and CD86 expression on ZsG+ DC1, but did not increase production of IL-12. These results suggested that Tregs may restrain CD8+ T cell priming by inhibiting DC1s – a hypothesis that was confirmed using ex vivo cultures of naive OT-I T cells, ZsG+ DC1s, and Tregs isolated from the tumor-draining mLN. Single-cell RNAseq further revealed that the presence of Tregs in co-cultures reduced expression of transcripts associated with effector functions, and increased those associated with inhibition of effector T cell differentiation and T cell fate decisions.
Comparing the suppressive capacities of Tregs from the mLN and iLN, the researchers found that Tregs from the mLN more potently inhibited CD25 and granzyme B expression on primed CD8+ T cells, and CD80, CD86, and (to a lesser extent) IL-12 on DC1s (regardless of the DC source), suggesting tissue-specific differences in the suppressive capacity of Tregs. When signals 2 and 3 were supplemented in the form of anti-CD28 (agonist) and IL-12, priming was effectively restored. This effect was recapitulated in vivo, where the administration of anti-CD28 and IL-12 during priming increased CD25 and granzyme B on tumor-reactive CD8+ T cells.
Using immunofluorescence imaging to investigating just how Tregs suppress DC1s and subsequent T cell priming, Zagorulya et al. found that in OT-I microniches – circular regions centered at OT-I T cell clusters that contained at least one DC1 – Tregs and DC1s were closer to one another in mLNs than in iLNs, while outside of OT-I microniches, they were farther apart. When MHC-II was blocked with antibodies or knocked out on DC1s, expression of CD80, CD86, and IL-12 increased on ZsG+ DC1s, and CD8+ T cell priming was restored, suggesting that Tregs suppress the priming capacity of DC1s through direct interactions with MHC-II on DCs.
To better understand why Tregs exerted more suppression in tumor-draining mLNs than tumor-draining iLNs, the researchers found that both the abundance of Tregs and the SIINFEKL-reactive CD8+ T cell:Treg ratios were comparable between locations. Paired RNA and TCRseq showed that Tregs in tdLNs, but not naive LNs, were enriched for a cluster of “activated c1” Tregs with increased clonal expansion. Within this Treg cluster, cells from the mLN expressed more transcripts associated with immunosuppression, IFN responses, and Th1 polarization, while those from the iLN cells were enriched for transcripts associated with T cell activation and Treg survival and stability. Among canonical Th1, Th2, and Th17-polarizing transcripts, the researchers identified high expression of Tbx21 (encoding T-bet), which is associated with the Th1 phenotype. Th1 polarization was further confirmed by showing that the canonical Th2 and Th17 transcription factors (Gata3 and RORγt, respectively) were not increased. Further, Tregs in the mLN expressed higher levels of Treg markers (NRP1, Helios), effector activation molecules (PD-1, CTLA-4), and suppressive molecules (NRP1, CTLA4, CD39, CD73), while Tregs in the iLN produced higher levels of TGFβ1 and CD25, suggesting distinct phenotypes and suppressive mechanisms between Tregs in each location.
While DC1s were found to be required for Treg polarization towards a Th1 phenotype, mediating increased T-bet and T-bet+CXCR3+ Th1-like effector Tregs, they did not account for tissue-specific differences in Treg polarizations. Instead, the researchers hypothesized that IFN signaling may play a role, as IFN transcripts were preferentially enriched in Tregs from mLNs. Testing this, the researchers showed through IFNγRknockout that IFN signaling was responsible for the induction of T-bet, CXCR3, and CD39 in both tdLNs, but that this program was predominantly induced in the mLN, where IFNγ was more abundant. This difference in IFNγ levels was also apparent in naive mice, but was equalized in germ-free mice, suggesting that the increase in IFNγ in the mLN was associated with the commensal microbes that are present in the lungs. Treg-specific ablation of IFNγ or transient blockade of IFNγ using antibodies administered early after tumor implantation effectively reduced Th1-like Tregs and restored CD8+ T cell priming in the mLN.
Finally, Zagorulyaet al. analyzed RNAseq data from TILs from human melanoma, and found that Treg expression of IFN response programs and either TBX21 or CXCR3 transcripts correlated with ICB response, while the CD8+/Treg ratio did not, suggesting that their findings in mice might also be relevant in patients.
Overall, these results suggest that increased IFNγ in the mLN, associated with the local lung tissue microbiome, polarizes Tregs towards a Th1-like effector phenotype dependent on the presence of DC1s. Through direct interactions with MHC-II on DC1s, these Th1-like Tregs suppressed signals 2 and 3 on DC1s, decreasing their capacity for effective CD8+ T cell priming. Strategies to disrupt this mechanism, including transient Treg depletion, IFNγ blockade, and supplementing signals 2 and 3 using anti-CD28 and IL-12 were each effective strategies in rescuing cytotoxic T cell responses, and could represent avenues of further exploration for improving antitumor immunity.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, lead author Maria Zagorulya answered our questions.

What was the most surprising finding of this study for you?
Increasing evidence suggests that the tissue site of tumor growth dictates the strength of antitumor immunity. However, the tissue-specific mechanisms of immunoregulation remain poorly understood. In this study, we found a surprising link between the baseline levels of interferon-gamma in tissue-specific lymph nodes and local immunosuppression. Specifically, interferon-gamma abundance in the lung tumor-draining lymph node drove the local induction of suppressive TH1-like effector regulatory T cells (Tregs). These suppressive Tregs potently restrained the stimulatory capacity of type-1 conventional dendritic cells (DC1), resulting in suboptimal T cell priming and the consequent induction of dysfunctional tumor-reactive CD8+ T cell responses.
What is the outlook?
Effective antitumor immunity requires the disruption of peripheral tolerance. Our finding that baseline levels of interferon-gamma vary across distinct tissue-specific lymph nodes and regulate Treg functional states suggests that local cytokines can control the tolerance setpoint in different contexts. However, the mechanisms that maintain the high levels of interferon-gamma in the lung-draining lymph node and thereby regulate the lung-specific tolerance setpoint remain unknown. Intriguingly, commensal bacteria were required for the observed lung lymph node-specific abundance of interferon-gamma, implicating local immune populations capable of responding to these microbes. Further studies are needed to understand the link between lung lymph node interferon-gamma levels and the microbiome.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently learned that cacti can survive snow! Coming from a cold climate, I had always associated the cactus with extreme heat. I was proven wrong on a trip to Arizona this past winter. I never thought snow-covered cacti existed, but there they were!



