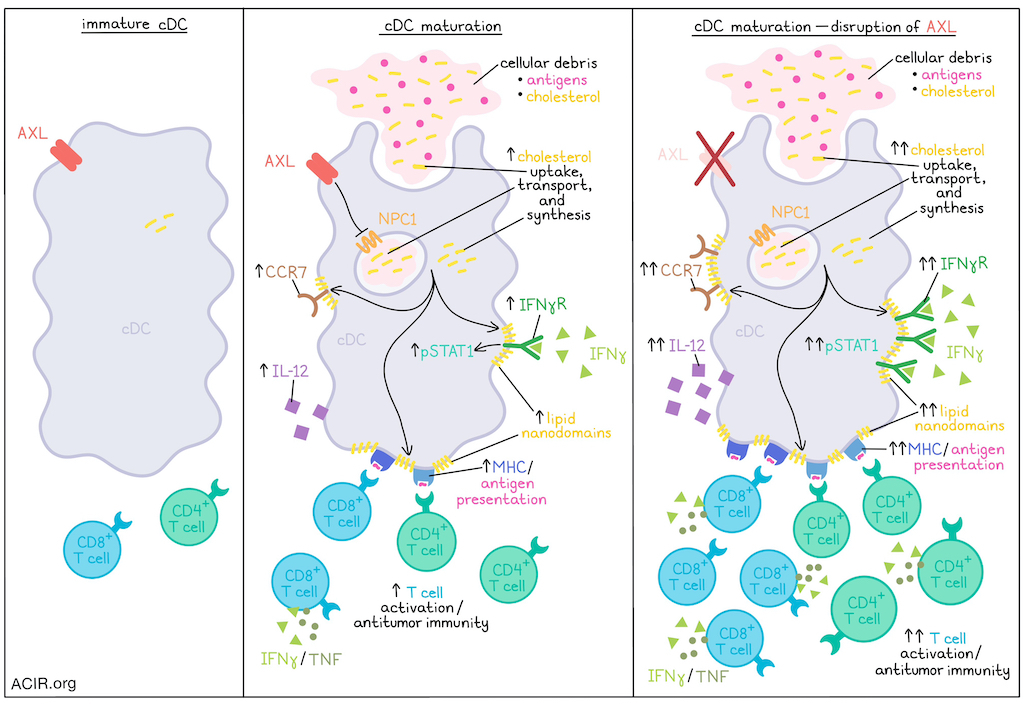
The maturation of dendritic cells plays a critical role in initiating a cascade of immune responses, but exactly how cDCs mature in the absence of inflammation is not entirely understood. Investigating this process, Belabed and Park et al. found that following the uptake of cellular debris and antigens, cDCs increase transport and synthesis of cholesterol, which supports the assembly of lipid nanodomains on cell membranes, allowing for the stabilization of maturation markers and receptors that support enhanced immune activation. In investigating this mechanism, the team also identified potential points of intervention that could be targeted to enhance antitumor immunity. Their results were recently published in Nature Immunology.
To begin, Belabed and Park et al. evaluated previously published transcriptional data for mature cDCs and found that, in addition to genes encoding molecules related to migration, co-stimulation, cytokine signaling, and chemokine signaling, mature cDCs were also marked by a signature of cholesterol synthesis and transport. To study this further, the researchers utilized a model in which naive cDCs were matured by feeding them debris from epithelial cells expressing BFP as a surrogate antigen. Labeling cholesterol with GFP-tagged probes, the researchers were able to quantify cholesterol in cell membranes, and found that it was increased in mature versus immature cDCs. Similar results were observed for CCR7+ (mature) compared to CCR7- (immature) cDCs isolated from digested tumor-bearing lungs. Interestingly, disrupting the reservoir of cholesterol (agonizing the LXR pathway to promote cholesterol efflux, treating with sivastatin to block cholesterol synthesis, or inhibiting cholesterol transporter NPC1) abrogated expression of maturation markers, including CCR7. Meanwhile, fueling cholesterol synthesis (treating with mevalonic acid) increased expression of maturation markers. Further, labeling the cellular debris fed to cDCs during maturation revealed that cholesterol derived from the debris was incorporated and mobilized in mature cDCs. However, maturation of cDCs induced through TLR2, TLR3 or TLR4 agonism was also dependent on cholesterol synthesis and transport, suggesting that cholesterol accumulation is essential for maturation, even in the absence of external sources.
Given that markers of maturation, including IFNγR and MHC-II, rely on stabilization by lipid nanodomains on the plasma membrane for signaling and function, the researchers marked lipid nanodomains with CTxB and found that they were increased in mature cDCs both in vitro and ex vivo. When cholesterol was removed or sequestered, lipid nanodomain formation and expression of maturation markers reduced. This effect was also found to be dependent on cholesterol transport by NPC1, suggesting that increased cholesterol supports lipid nanodomain formation and maturation marker expression.
Next, the researchers hypothesized that surface sensors associated with the uptake of cellular debris might regulate cholesterol transport and cDC maturation. While most phagocytic receptors were downregulated following antigen uptake, the receptor tyrosine kinase AXL was found to be maintained on mature cDCs. Using scRNAseq to profile MHC-II+CD11c+ cells from the lungs of wild-type (Axl+/+) and Axl-knockout (Axl−/−) mice, the researchers found that the mature cDCs from Axl−/− mice upregulated expression of Npc1 and other mRNAs encoding enzymes involved in de novo cholesterol synthesis, suggesting that AXL may regulate these pathways upon uptake of debris in in wild-type cDCs. In vitro, Axl−/− mature cDCs showed increases in membranous free cholesterol, transport of cholesterol derived from debris to the plasma membrane, and expression of maturation markers compared to wild-type cDCs, further supporting the hypothesis that AXL acts as a checkpoint on cDC maturation. Evaluating knockouts of AXL and NPC1 alone or in combination, the researchers confirmed that AXL acted as a checkpoint on NPC1, suppressing cholesterol transport and synthesis. Imaging of lipid nanodomains in Axl-/- mature cDCs with high IFNγR and MHC-II expression also showed increased surface area occupied by nanodomains, increased pSTAT1 (indicative of increased IFNγR signaling), and increased IL-12 production.
Investigating the antigen-presenting capacity of mature cDCs under different circumstances, Belabed and Park et al. utilized OVA as a model antigen and evaluated activation of OT-I and OT-II T cells. This showed that when the cholesterol reservoir was disrupted, cDCs had a lower capacity for T cell activation. On the other hand, AXL-knockout cDCs had an enhanced capacity for T cell activation, dependent on cholesterol synthesis and transport.
To study these effects in vivo, the team intravenously injected Npc1+/+ and Npc1+/− mice with 4T1 breast cancer cells to model accumulating debris in the lungs. Ex vivo coculture of mature cDCs showed that those from Npc1+/− mice were less capable of activating T cells, and those from lung metastases showed reduced expression of maturation markers than those from Npc1+/+ mice. In Axl-/- mice, T cells showed greater activation potential (IFNγ+ and TNF+) than those in Axl+/+ mice.
Based on the relatively restricted expression of AXL to mature cDCs, the researchers investigated pharmacological inhibition of AXL using bemcentinib, and found that it reduced tumor loads in treated mice. Given that AXL is also expressed in tumor cells and is associated with tumor cell invasiveness, the researchers also evaluated tumor growth in Axl-/- mice and mice with deletion of Axl specifically in DCs, and found that tumor growth was reduced in these settings as well, suggesting that the effects of bemcentinib were not solely due to direct effects on tumor cells. Both Axl−/− mature cDCs and cDCs treated with bemcentinib showed enhanced assembly of lipid nanodomains on their cell surface, produced more IL-12, and activated more T cells expressing effector cytokines (IFNγ and TNF). Depletion of either CD8+ or CD4+ T cells abrogated the therapeutic advantage of AXL deficiency, with CD4+ T cells having a stronger impact. NPC1 inhibition also abrogated the effects of AXL deficiency.
Finally, the researchers used an in vitro culture system in which immature cDCs derived from human cord blood were fed to induce maturation. Like in the murine models, this induced upregulation of genes encoding enzymes involved in de novo cholesterol synthesis, AXL, and classical markers of maturation. NPC1 inhibition reduced the expression of maturation markers, cross-presentation of a model antigen (NY-ESO-1) from apoptotic debris, and activation of antigen-specific CD8+ T cells, suggesting that the findings in mouse models indeed translated to human cDCs.
Overall, these results unveil a mechanism in which antigen uptake or receptor-mediated activation of cDCs increases cholesterol synthesis and transport, supporting the assembly of lipid nanodomains and the expression of cDC maturation markers. This enhanced maturation was associated with increased antigen presentation and activation of T cell responses that could mediate antitumor immunity. Importantly, AXL was found to regulate this mechanism through inhibition of NPC1-mediated cholesterol transport, and inhibition of AXL emerged as a potential target for cancer immunotherapy.
Write-up and image by Lauren Hitchings



