T cell therapies have been used to treat numerous cancers, but they are often limited by poor proliferation, limited persistence, immunosuppressive tumor environments, and exhaustion. In order to overcome some of these limitations, Garcia and Daniels et al. investigated naturally occurring mutations in T cell neoplasms (including clonal cells from autoinflammatory conditions and T cell lymphomas) that allow these abnormal cells to survive and thrive. The researchers then exploited these mutations in CAR and TCR T cell therapies to show how they could be used to improve antitumor efficacy. Their results were recently published in Nature.
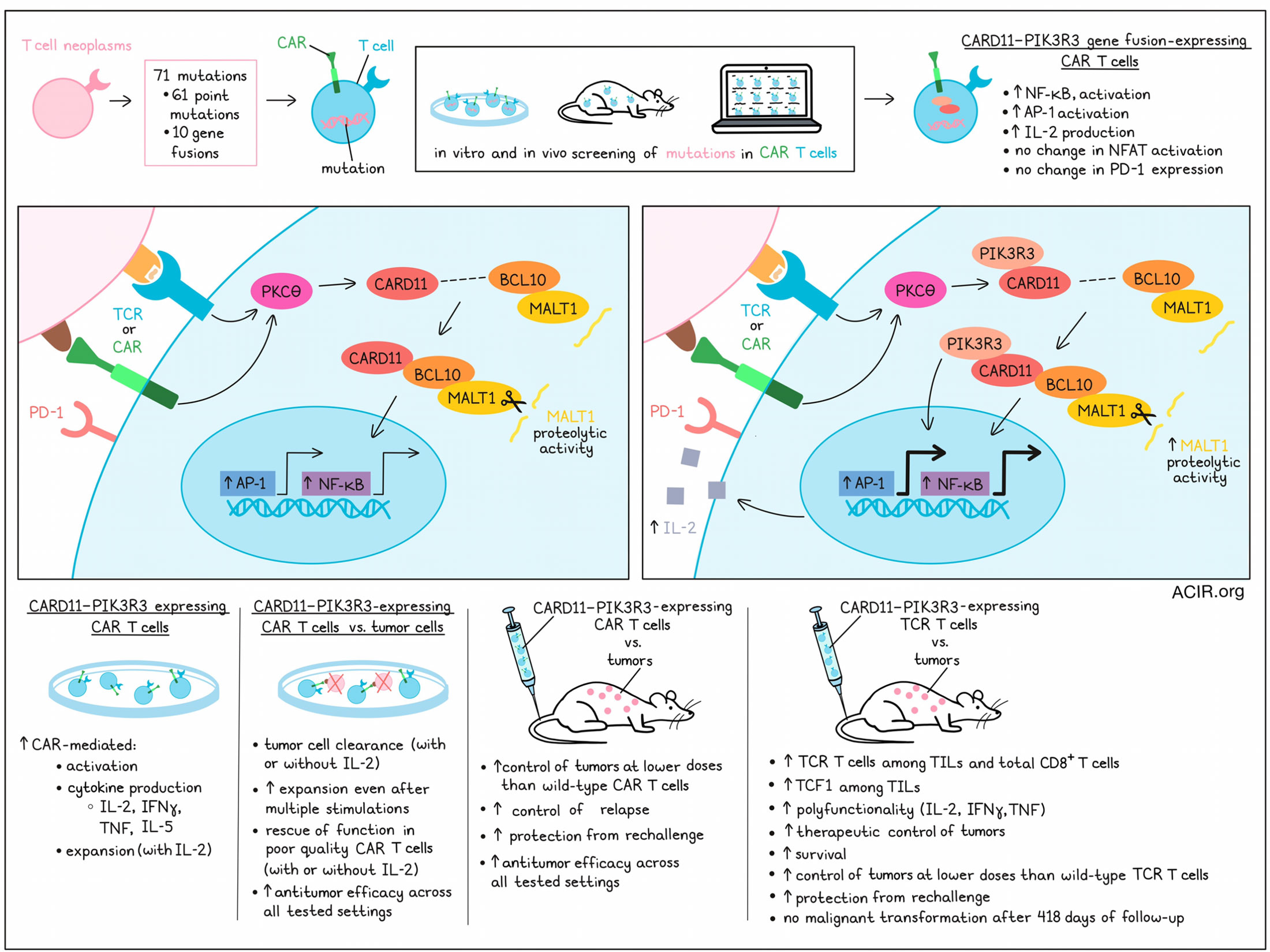
Garcia and Daniels et al. began by identifying 71 mutations in possible genetic drivers of T cell abnormalities, including 61 point mutations and 10 gene fusions. They then introduced these mutations, along with a CAR (CD19-CD28z-CAR or a CD19-BBz-CAR) vector, into triple-reporter Jurkat T cells for NF-κB, AP-1, and NFAT pathway activation. Measuring activation of these 3 pathways, as well as production of IL-2 and expression of PD-1 in vitro, the researchers identified numerous mutations with significant and diverse impacts on T cells, some of which were more pronounced when the target antigen was present.
Next, the mutations that induced changes were screened in primary human CAR T cells. For this experiment, transferred cells were pooled, sorted, and transplanted into an immunodeficient xenograft model in which CAR T efficacy is usually limited. Taking note of constructs that were depleted or enriched, and investigating them in vitro, the researchers noticed that the fusion of CARD11 and PIK3R3 stood out for markedly enhancing NF-κB and AP-1 signaling and IL-2 production, without increasing NFAT or PD-1 – a pattern associated with reduced T cell dysfunction and stronger effector and memory functions. This fusion was originally identified in a single patient with CD4+ cutaneous T cell lymphoma.
In normal T cells, TCR signaling activates PKCθ, which allows CARD11 to recruit BCL10 and MALT1, forming the CARD11–BCL10–MALT1 (CBM) complex. The CBM complex then goes on to mediate enhanced NF-κB and AP-1 transcriptional activity, as well as MALT1 proteolytic activity. When the CARD11–PIK3R3 gene fusion mutation was introduced, NF-κB signaling was still dependent on BCL10 binding, and MALT1 activity was maintained, suggesting that the CBM complex still formed and was functional. Introduction of an alteration in the phospho-tyrosine binding pocket of the PIK3R3 SH2 region significantly reduced NF-κB signaling, suggesting that CARD11–PIK3R3 maintains CBM signaling and further enhances it through PIK3R3 SH2-dependent bindings.
Looking at the functionality of CARD11–PIK3R3-expressing T cells, the researchers noted that CD4+ T cells were enriched for gene signatures related to cell cycle, while CD8+ T cells were enriched signatures related to RNA metabolism, cytokine signaling, and translation. Further, while CARD11–PIK3R3-expression was not sufficient to induce a fully activated phenotype on its own, it did enhance CAR-mediated activation, expansion (in the presence of IL-2), and secretion of IL-2, IFNγ, TNF, and the TH2 cytokine IL-5, particularly in CD8+ T cells.
In co-culture experiments, CAR T cells expressing CARD11–PIK3R3 cleared target cells, even in absence of supplementary IL-2, suggesting potential efficacy in immunosuppressive tumor microenvironments, where IL-2 is in short supply. They also showed evidence of increased expansion after multiple stimulations, suggesting greater antigen-dependent proliferative potential and resistance to exhaustion. Further, where CAR T cells derived from one donor failed to fully eliminate tumors (even with IL-2), the addition of CARD11–PIK3R3 enabled tumor cell clearance with or without IL-2, suggesting that CARD11–PIK3R3 could rescue the functionality of otherwise poor-quality CAR T cells. Overall, the inclusion of CARD11–PIK3R3 in CAR T cells consistently improved antitumor efficacy across every test setting.
Moving to mouse models, Garcia and Daniels et al. evaluated CARD11–PIK3R3-expressing CD19-targeted CAR T cells in a CD19+ Nalm6 xenograft leukemia model. In this setting, the leukemia was controlled, but the mice experienced lethal weight loss, likely due to GvHD. Knocking out the native TCR eliminated this toxicity, allowing for 100% survival at day 33, and control of leukemia at doses lower than would be required with wild-type CAR T cells. At higher doses, CARD11–PIK3R3-expressing CAR T cells were still well tolerated, could prevent relapse, and protected mice from rechallenge better than wild-type CAR T cells. CARD11–PIK3R3-expressing cells also outperformed wild-type CAR T cells in both a subcutaneous mesothelioma model and a CD19+ B16F10 melanoma model, suggesting potential efficacy in solid tumors and immunosuppressive tumor microenvironments, even without lymphodepletion.
Next, the researchers evaluated whether CARD11–PIK3R3 could also enhance the antitumor efficacy of TCR T cells. Using a B16F10-OVA model, the researchers transferred a mix of fluorescently tagged OT-I T cells, 10% of which were carrying the CARD11–PIK3R3 construct. A week after transfer, CARD11–PIK3R3-expressing cells increased 145-fold among TILs compared to control cells. Among total CD8+ T cells, there was a significantly increased fraction of transferred cells, even without normalization, and 4 of 5 tumors were cleared by day 21. Similar observations were made in transduced pmel-1 T cells. Further analysis of these models revealed that BCL10 binding was required for antitumor efficacy, and that TCF1 (associated with stemness) was increased among TILs, but not T cells in spleens or tdLNs. CARD11–PIK3R3 expression also increased polyfunctionality, increasing the production of TNF, IFNγ and IL-2 both individually and collectively.
Evaluating antitumor efficacy in a more advanced therapeutic setting, the researchers administered OT-I T cells 12 days after tumor inoculation, without preconditioning, and found that CARD11–PIK3R3 expression significantly enhanced tumor control and prolonged survival, with 3 of 5 mice tumor-free at more than 3 months – a time point at which no control or OT-I-treated mice survived. Overall, treatment was well tolerated, though one mouse died with a skin ulceration. In a cell dose–response experiment, CARD11–PIK3R3 enabled superior tumor control at both a 20-fold and 100-fold lower dose than control cells, without lymphodepletion, and protected mice from rechallenge. Mice from this study were followed up to 418 days after T cell transfer, and no evidence of malignant transformation was observed. Similar results were observed in a human TCR T cell model.
Overall, these results show that CARD11–PIK3R3 enhanced CBM signaling, in turn, enhancing the antitumor efficacy of both CAR and TCR T cells in numerous settings. The inclusion of CARD11–PIK3R3 in T cell therapies could be used to improve treatments and overcome various hurdles, including poor T cell quality, immunosuppression, and the requirement for lymphodepletion.
Write-up and image by Lauren Hitchings



