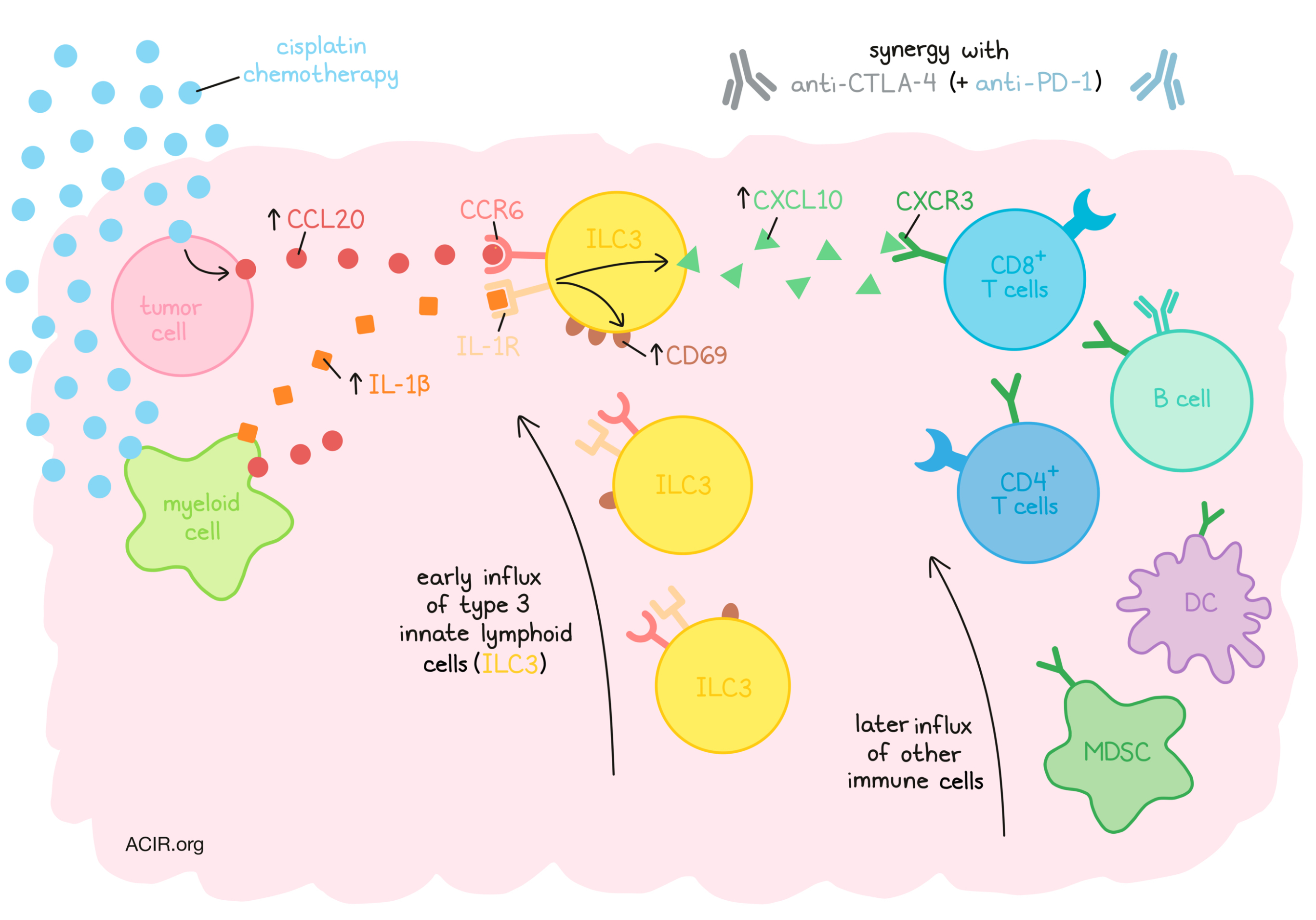
Immunologically cold tumors generally respond poorly to immune checkpoint blockade (ICB). Defining the mechanisms that can increase immune infiltration into these tumors is essential to improving ICB therapy efficacy. Cisplatin chemotherapy treatment has been shown to synergize with ICB by increasing immune cell infiltration in cold tumors, but the mechanisms behind this remain unclear. Therefore, Bruchard et al. set out to answer this question using various animal models. Their results were recently published in Nature Immunology.
The researchers made use of various cold tumor models, including the TC-1 transplantable lung, 4T1 transplantable triple-negative breast cancer, and the carcinogen urethane-induced lung cancer murine models, with a primary focus on the TC-1 model. These tumors are characterized by low immune cell infiltration and resistance to ICB. Cisplatin treatment increased infiltration of type 3 innate lymphoid cells (ILC3s) at day 1 after treatment, later followed by other immune cell types. Most of these ILC3s expressed CCR6, but not NKp46 (an activating receptor found on NK cells). Treatment with carboplatin had similar effects, while treatment with oxaliplatin did not, suggesting various platinum-based chemotherapeutics had different impacts on immune infiltration. The antitumor effects of cisplatin and carboplatin were reduced when immunodeficient mice were studied, while the antitumor effects of oxaliplatin were not affected. Pooling data from several tumor models showed a clear correlation between ILC3 accumulation and T cell, B cell, DC, and myeloid-derived suppressor cell infiltration after cisplatin treatment.
To assess whether ILC3s were essential for the antitumor effects of cisplatin, the researchers assessed the response to treatment in the presence and absence of these cells. Since ILC3s express RORyt, the inhibitor digoxin could be used to deplete these cells. This inhibitor also depletes Th17 cells, so the researchers compared assays with the specific blockade of Th17 cells alone to assess ILC3-specific effects. Digoxin reduced the antitumor and immune infiltration effects of cisplatin, which were shown to be ILC3-specific effects. These results were confirmed in models devoid of ILC3s or Th17 cells, suggesting ILC3s were essential for cisplatin’s antitumor effects.
Next, Bruchard et al. assessed the role of ILC3s in the efficacy of the combination of cisplatin and ICB. They assessed anti-CTLA-4, as it is the most expressed checkpoint on T cells in the TC-1 model. The combination was synergistic, while anti-CTLA-4 alone had no effects. The synergistic effect was only observed when ILC3s were present in the model, while Th17 depletion had no apparent impact on efficacy. In the “hot” tumor model MC38, which is very responsive to anti-CTLA-4 and anti-PD-1 (dual) ICB, depletion of ILC3s also reduced efficacy, while Th17 depletion did not.
To determine what role the ILC3s play in the attraction of other immune subsets, single-cell RNAseq was conducted on sorted ILCs from TC-1 tumors. After cisplatin treatment, ILC3s expressed high levels of CXCL10, known for its role in lymphoid cell recruitment. Untreated ILC3s could produce CXCL10 protein, and cisplatin treatment increased this production. To assess the role of this chemokine, an antibody targeting the receptor of CXCL10, CXCR3, was used in vivo. This blockade reduced the antitumor effects of cisplatin, and the addition of digoxin did not further inhibit this. It also reduced immune infiltration, while the ILC3 population was not affected. This suggests that CXCL10 plays an essential role in the effects ILC3s have in response to cisplatin treatment. Without anti-CXCR3 and in the presence of ILC3s, cisplatin caused an accumulation of CD4+ and CD8+ T cells, with increased proportions of activated and proliferating T cells expressing granzyme B, IFNγ, TNF, and IL-2.
The researchers found that the early influx of ILC3s after cisplatin treatment was not due to the increased proliferation of CCR6+ ILC3s. There was, however, an increase in CCL20 expression in the tumor microenvironment due to the tumoral production of this chemotactic ligand of CCR6. Again, only cisplatin and carboplatin had this effect on tumor cells; oxaliplatin did not, suggesting it may be a factor in the ILC3 increase after treatment. Indeed, depletion of CCL20 reduced the number of CCR6+ ILC3s and T cells, as well as the efficacy of treatment. Additionally, in “hot” MC38 tumors, both tumor and myeloid cells produced CCL20 after dual ICB, and depletion of this chemokine reduced the ILC3 and T cell populations, reducing treatment efficacy.
Some known activators of ILC3s are IL-23 and IL-1β. After cisplatin treatment, the levels of IL-23 in the tumor remained stable, while IL-1β increased. Gene enrichment assessment of ILC3 RNAseq data showed an enrichment of the IL-1R module after cisplatin treatment. Myeloid cells produced IL-1β after treatment, and the use of the soluble receptor IL-1RA, which inhibits IL-1β, resulted in reduced activation and accumulation of ILC3s and their production of CXCL10. It also reduced T cell infiltration and the antitumor effects of cisplatin. Because IL-1β is also known for its role in γδ T cells activation, the researchers confirmed that these cells did not play a role in the cisplatin-induced effects.
In vitro stimulation of ILC3s obtained from tumors with IL-1β or IL-1β and IL-23 activated the cells, resulting in higher CD69 expression and CXCL10 production. In untreated MC38 tumors, a higher concentration of IL-1β was found than in untreated TC1 tumors, and inhibition of this cytokine resulted in reduced intratumoral T cell infiltration and efficacy of dual ICB.
Finally, to assess the role of CCL20 and IL-1β, the TC-1 model was treated with CCL20 and IL-1β intratumoral injections, resulting in an increase in the frequency and activation of CCR6+ ILC3s, increased T cell influx, and inhibited tumor growth, which could be counteracted with digoxin. In MC38 tumors, adding these cytokines also reduced tumor growth and improved response to dual ICB, and digoxin treatment canceled these effects.
These data demonstrate that the production of CCL20 and IL-1β in the TME in response to cisplatin treatment leads to the early recruitment of ILC3s to the tumor. The production of CXCL10 by these ILC3s then attracts T cells to the TME, turning cold tumors hot and synergizing with ICB treatment. It will be of interest to determine whether this effect can also be observed after chemotherapy treatment in patients with cold tumor types that respond poorly to ICB.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Mélanie Bruchard and lead author François Ghiringhelli answered our questions.

What prompted you to address this research question?
Immunotherapy is one of the leading treatments in modern oncology. Despite this, these treatments lack effectiveness in many cases. The association with chemotherapy, such as cisplatin, increases its efficiency, but the mechanism of action is not yet clear. Studies have shown that some chemotherapies can activate cells of the adaptive immune response via the immunogenic death mechanism. For cisplatin, things seem to be more complex, because this drug does not induce this immunogenic death mechanism, but an important immune recruitment to the tumor can still be observed. Considering ILC3 are essential to the genesis of lymph nodes during fetal life, and are therefore implicated in the recruitment of immune cells, we then formulated the hypothesis that innate lymphoid cells could have a role in the immune effect of chemotherapies.
What was the most surprising finding of this study for you?
It was the first demonstration of the role of innate lymphoid cells of group 3 in the response to chemotherapy. Thus, these cells play an essential role in the recruitment of immune cells within the tumor tissue, which is surprising considering their very small number (less than 0.5% of the tumor). These data suggest that there is a broader interest in understanding the role of different innate lymphoid cells in the effect of immunotherapies and chemoimmunotherapies.
What is the most interesting thing you have learned recently outside of the lab?
MB: I recently learned that the area around Chernobyl is now a sanctuary for many species (bears, wolves, wild horses, bisons...). As humans have left, nature has reclaimed its rights, making room for wildlife. Moreover, scientists have now installed cameras to follow and study these animals. I think it's a really nice way to make some good out of this disaster.
FG: Outside of the lab I am a physician and treat cancer patients. It's really cool to be able to talk to patients about this research. It's a way to give hope. With my kids it's more fun, at school and when we get guests, they like to say that I'm not a real doctor but the mouse doctor.



