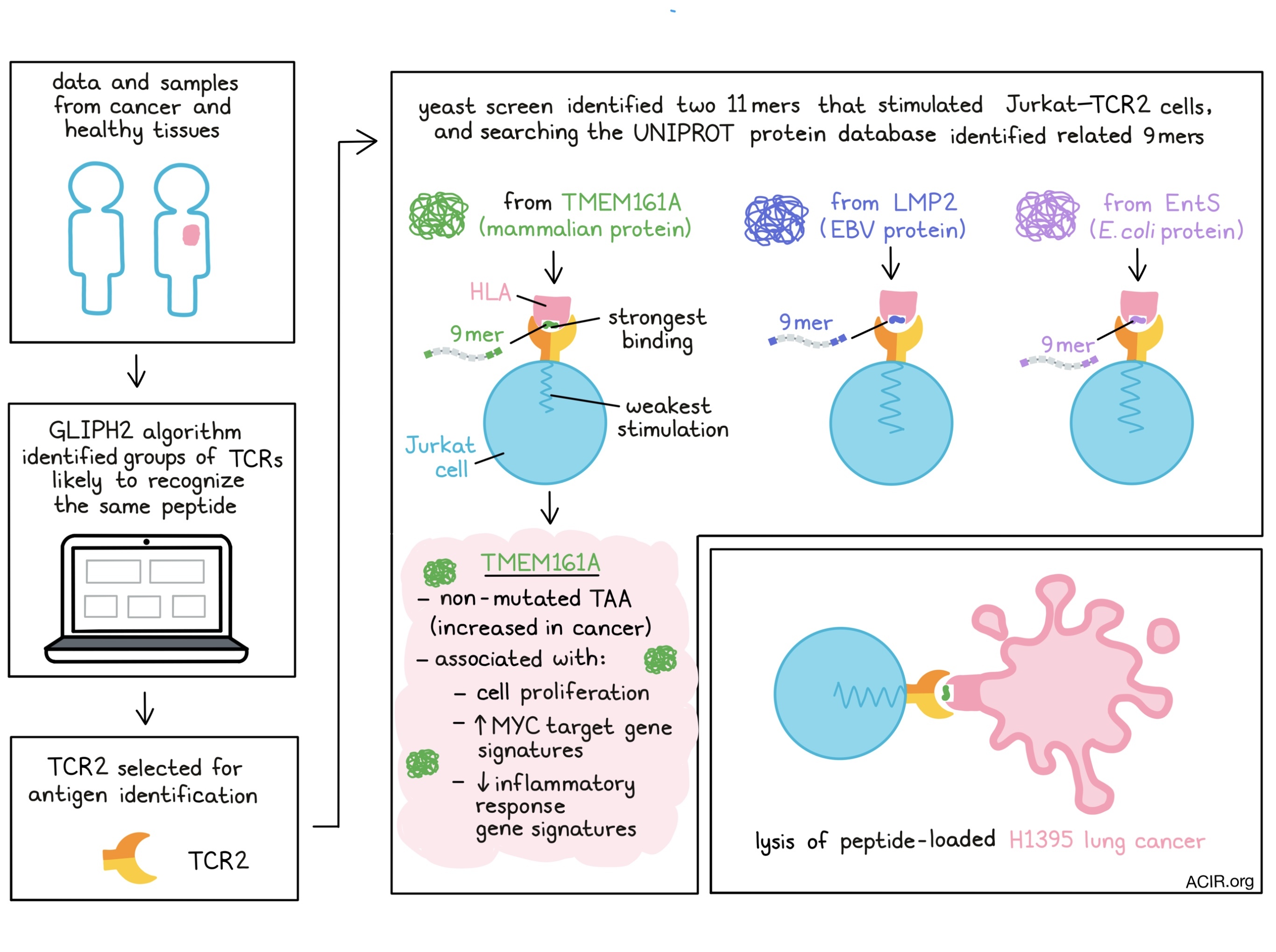
Virus-specific tumor-infiltrating T cells have previously been considered bystander cells not contributing to the antitumor response in the same way that T cells specific to tumor-associated antigens (TAAs) might. To begin to unpack the vast amount of TCR sequence data available from tumor and normal tissue samples, Chiou and Tseng et al. used the algorithm GLIPH2 to identify groups of TCRs with similar antigen specificities in a large cohort of patients with non-small cell lung cancer (NSCLC), uncovering cross-specificities between TAA and pathogen antigens. Their data were recently published in Immunity.
The GLIPH2 algorithm was used on a recently published dataset of 778,938 distinct CDR3β sequences obtained from tumor and healthy adjacent tissue from 178 patients with NSCLC. The algorithm clusters TCR sequences into shared specificity groups likely to recognize the same peptide amino acid sequence presented by HLA. In this dataset, 66,094 shared specificity groups were discovered, of which 4,226 showed evidence of clonal expansion, and 435 were enriched in the tumor tissue.
The researchers validated these data by adding CDR3β sequences (from an available HLA tetramer database) with viral specificities to their dataset and rerunning GLIPH2 to annotate specificity groups based on shared CDR3β-specificity. The annotated specificity groups were clustered into communities using network analysis. The majority of antigen specificity groups with distinct sequences shared HLA-restricted specificity to common pathogens (Influenza, CMV, EBV).
In the lung cancer dataset, only an average of ~0.4% of the TCR repertoire was shared between two patients. When looking at the shared specificity groups, however, that percentage increased to 5.3%. Using statistical bootstrapping, the researchers found that the number of HLA-A*02:01-enriched specificity groups reached saturation at ~70 patients, suggesting that a complete set of TCR specificity groups could be established with a limited dataset.
The researchers analyzed tumor tissue samples from 15 patients with early-stage NSCLC using single-cell TCR sequencing. The 4,704 paired CDR3α and CDR3β sequences found were combined with the earlier obtained dataset and entered into GLIPH2. Four of the detected virus-specific T cell clones were validated by engineering Jurkat76 cells to express the TCRs, and then co-culturing these cells with HLA-A*02+ T2 cells pulsed with the viral peptides. Three Jurkat-TCR cells responded to the predicted antigens, confirming GLIPH2’s predictive value.
The researchers then focused on the tumor-enriched specificity groups that had a paired TCRα/β clonotype and were significantly enriched with HLA-A*02 alleles. One group of interest was the specificity group with the “S%DGMNTE” CDR3β motif (% represents a variable AA). From this group, a TCRα/β clonotype (named TCR2) was selected for antigen identification.
Using a yeast library displaying peptides of four different lengths in the context of HLA-A*02:01, cognate ‘mimotopes’ for TCR2 were identified, and the top 20 were used in an in vitro stimulation assay. The top 2 peptides (both 11mers) stimulated the TCR2-expressing Jurkat cells. Searching the UNIPROT protein database identified 9mers that were closely related to the mammalian protein TMEM161A, the EBV protein LMP2, and the E. Coli protein EntS, all of which stimulated Jurkat-TCR2 cells. These data were further confirmed using the full-length proteins in HLA-A*02+ 293T cells, which also stimulated the TCR2-T cells. TMEM161A was the weakest in eliciting a response, but, counterintuitively, had the most stable binding affinity. These data suggest that these T cells were cross-reactive to viral and epithelial antigens.
Moving back to tissue sections, the researchers found increased expression of the TMEM161A protein in human lung cancer compared to healthy lung tissue, and the TCGA NSCLC dataset also showed higher levels of the TMEM161A transcript in tumors, with the highest expression in squamous cell carcinomas. Whole exome sequencing of specimens from the original cohort did not identify a mutation in this locus, suggesting the protein is a non-mutated TAA; this was confirmed in the TCGA pan-lung cancer dataset. TMEM161A expression was associated with cell proliferation and MYC target gene signatures, and was inversely correlated with inflammatory response gene signatures. In the NSCLC cohort, 40% of the HLA-A*02+ patients had TMEM161A-specific T cells, and T cells with the “S%DGMNTE” motif were more common in squamous cell carcinomas.
To further characterize the T cells that were cross-reactive to TMEM161A and EntS, tetramers were used to sort T cells from the blood of healthy donors and patients with NSCLC. Cross-reactive cells were found in a similar frequency in healthy donors and patients, and single-cell sequencing revealed they were mostly effector T cells.
The sorted cells were functionally validated by generating Jurkat cells expressing the TCRα/β chains identified with the tetramer-sort, and assessing the reactivity to TMEM and pathogen-derived 9mers in the context of HLA-A*02:01. The “S%DGMNTE” CDR3β motif responded to all cross-reactive peptides when paired with the TCR2α chain. Furthermore, co-cultures of the peptide-loaded lung cancer cell line H1395 with TCR2-T cells showed target cell lysis, again with a weaker response to the 9mer for TMEM, perhaps reflecting the attenuated response to chronically expressed TAAs in progressing tumors.
The researchers then performed single-cell transcriptome and TCR sequencing on sorted T cells from ten patients with NSCLC to assess cell states. The most expanded clusters had effector and resident memory phenotypes. The tumor-enriched specificity groups were mostly effector cells and expressed EOMES, KLRG1, and GZMK. Pseudotime analysis indicated that these cells adopted distinct cell states. Virus-specific T cells, on the other hand, mainly had effector and tissue resident-memory phenotypes.
Finally, to assess these T cells' impact on checkpoint therapy, the TCR repertoires of two patients with NSCLC who responded to treatment were sequenced in pre- and post-treatment blood samples. Tetramer-stained T cell CDR3β sequences were used to annotate post-treatment expanded CDR3β clonotypes. Eleven clonotypes were found to contain three expanded CDR3β clones inferred to recognize viral antigens, which was confirmed with the Jurkat assay. EBV-specificity groups were expanded in the tumor compared to adjacent tissue, indicating that these cells may play a role in the response to checkpoint blockade. The EBV-specific clone TCR15 was inferred to have the same antigen specificity as two previously reported clones found in patients with response to checkpoint blockade.
The presented data shine new light on the cross-specificity of TIL between human and pathogen proteins and its role in immune checkpoint blockade. Characterization of TCRs using these methods will be of interest in other cancer types to understand the origin and dynamics of tumor-reactive T cells and reveal new antigens of interest to target with immunotherapy.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, we asked first co-authors Shin-Heng Chiou and Diane Tseng to answer our 3 questions.

What prompted you to tackle this research question?
SHC: The practical reason was that I was a trainee working in Dr. Davis's group when we started this project. Dr. Davis has a long-standing interest in understanding T cell specificity and I simply "married" this strong expertise of his with my focus in cancer.
DT: I regularly treat patients with lung cancer using checkpoint immunotherapies, but there is still so much to learn about how T cells recognize cancer and what they are “seeing”. I was very excited to apply novel computational and synthetic biology tools to better understand how the adaptive immune system recognizes cancer.
What was the most surprising finding of this study for you?
SHC: It was definitely the moment when I learned that the weakest agonist peptide from the tumor, antigen TMEM161A, actually bound TCR2 with the highest affinity. Before this, I was leaning towards the general consensus that strong agonist peptides usually have strong binding affinities to the cognate TCRs, and obviously there are exceptions.
DT: It was the finding that some T cells in tumors cross-react to recognize pathogen-derived antigens and tumor antigens. This helped me better understand at a mechanistic level how immune recognition of pathogens may shape antitumor immunity.
What was the coolest thing you’ve learned (about) recently outside of work?
SHC: I recently moved from the San Francisco Bay Area to Princeton for my new career. On one cold, rainy day, I discovered that there are salamanders in our backyard! I know this might sound a bit random, but back in Taiwan (where I originally came from), we needed to hike up to 3,500 meters above sea level to find salamanders.
DT: I recently moved to Seattle, WA and I’ve really enjoyed hiking Mt. Rainier. It was here that I saw for the first time the beautiful wildflower Castilleja, a species of Indian paintbrush



